
Angelman syndrome что это
Обновлено: 05.07.2024
Синдром Ангельмана – хромосомная аномалия, обусловленная мутацией генов и проявляющаяся у маленьких детей нарушениями психоэмоционального и физического развития. Больные беспричинно улыбаются и смеются, хаотично двигают верхними конечностями, страдают бессонницей. Внешне такие дети имеют добродушный и счастливый вид. Подобные отклонения заложены в организме ребенка еще до рождения.
Первым заболевание описал детский врач из Англии Гарри Ангельман в 1965 году, благодаря которому оно и получило свое наименование. Гарри наблюдал за больными детьми с практически одинаковой клинической симптоматикой и назвал их кукольными. Эти дети постоянно улыбались и казались счастливыми. На самом деле это не так. Улыбка – всего лишь гримаса, за которой скрывается несчастная человеческая душа.
дети с синдромом Ангельмана
Синдром Ангельмана имеет несколько равнозначных наименований: синдром «петрушки», синдром «счастливой куклы», синдром «марионетки», синдром «кукольных детей». Данный недуг характеризуется отсутствием некоторых генов в 15-й хромосоме или их мутацией. Синдром встречается одинаково часто как среди мальчиков, так и среди девочек. Это довольно редкий недуг: он возникает у 1 из 10000 новорожденных.
Поскольку заболевание генетическое, лечению оно не поддается. В настоящее время разработаны процедуры, способные улучшить качество жизни пациентов с данным диагнозом. Это занятия с логопедом и дефектологом, физиотерапия, медикаментозное воздействие. Если 15 хромосома повреждена незначительно, больные дети учатся говорить и самостоятельно себя обслуживать. Правильный уход и забота близких позволяют дожить больным детям до зрелых лет.
Симптомы
Первые признаки синдрома Ангельмана появляются к концу первого года жизни ребенка. Более явными симптомы становятся по достижению двухлетнего возраста. Синдром характеризуется полиморфностью клинических проявлений. Больных можно узнать сразу. Такие дети имеют характерный внешний вид.
- Особенности внешнего вида больного ребенка: несоответствие маленькой головы и нормального туловища, большой рот, постоянная улыбка, широкие межзубные промежутки, узкие губы, высунутый широкий язык, неправильный прикус, плоский затылок, гладкие ладони, заостренный подбородок, гипопигментированная кожа, стробизм, сколиоз, ходьба на прямых ногах.
- Нарушение психоэмоционального развития проявляется отставанием в умственном развитии, избыточной суетливостью, дружелюбностью, беспричинным смехом, эйфорией.
- Неврологические симптомы: тремор конечностей, атаксия, дискоординация движений, потеря равновесия, гипотония мышц, расстройство сна, истерия, нарушение речи, гиперактивность и гипервозбудимость, сложности с обучением.
- Двигательные расстройства: слабый контроль за движением языка и его беспричинное высовывание, трудности с глотанием и сосанием, поднятые или согнутые во время ходьбы руки, частое слюнотечение, неуемная жажда, излишне активные жевательные движения, нарушение мелкой моторики, эпилепсия.
С возрастом клиническая картина болезни значительно изменяется. Судороги и эпиприступы возникают все реже или исчезают совсем. Пациенты становятся более спокойными, у них налаживается сон. Мужчины и женщины с данным синдромом имеют моложавую внешность, скрывающую их истинный возраст. Половое созревание у них происходит вовремя, возможно даже рождение детей.

Поскольку заболевание является врожденным, все перечисленные аномалии могут проявиться сразу после родов. Современные методы диагностики позволяют выявить синдром внутриутробно и предотвратить рождение больного ребенка.
Видео: примеры детей с синдромом Ангельмана
Причины
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме материнского набора, но его характер и способ возникновения могут различаться. Иногда заболевание дебютирует в результате передачи измененной генетической информации от родителя, иногда является последствием спонтанных нарушений в геноме. Хромосомные аномалии удается определить примерно у 85-88% больных. Причиной синдрома может быть:
- Делеция. При данном дефекте часть генетического материала теряется или инактивируется. У 70% пациентов диагностируются обширные делеции области 15q12 хромосомы, в которой локализован активатор гена.
- Однородительская дисомия. ОРД определяется в 2-3% случаев болезни. В хромосомном наборе присутствуют две копии 15 хромосомы отца. Материнской хромосомы нет, ген также отсутствует.
- Дефект запечатления. Суть аномалии заключается в том, что центр запечатления, регулирующий активность локуса UBЕ3A, оказывается нефункциональным, «выключенным». Ген остается структурно целым, но не выполняет своих функций. Распространенность ДЗ – 3-5%.
- МутацияUBE3A. У 5-10% пациентов причиной болезни являются мутационные изменения гена. Они представлены инверсиями, микроделециями, транслокациями и дупликациями.
Диагностика и лечение синдрома Рубинштейна-Тейби
Для выявления синдрома Рубинштейна-Тейби используют данные общего осмотра больного, рентгенологических исследований и молекулярно-генетических анализов, вспомогательную роль могут играть УЗИ и МРТ. При осмотре определяются характерные для этого заболевания аномалии развития лица (изменение формы и размеров черепа и носа, антимонголоидный разрез глаз), укорочение и расширение фаланг больших пальцев на руках и ногах, иногда – полидактилия. У взрослых больных синдромом Рубинштейна-Тейби также выявляют уменьшение роста, глубокую умственную отсталость, искривления позвоночника. На рентгенограммах можно увидеть изменения костей фаланг, позвоночника и грудной клетки, костный возраст несколько отстает от фактического.
Молекулярно-генетическая диагностика синдрома Рубинштейна-Тейби выполняется врачом-генетиком, который может использовать множество методов для определения этого заболевания. Точечные мутации в генах CREBBP и ЕР300 выявляются посредством прямого автоматического секвенирования кодирующей последовательности. Методика FISH используется в том случае, когда подозревается наличие делеций или транслокаций на 16-й хромосоме, также приводящих к развитию синдрома Рубинштейна-Тейби. Так как на основании фенотипических данных крайне сложно определить возможный тип генетического дефекта, в рамках диагностики этого заболевания нередко приходится применять сразу несколько техник современной молекулярной генетики.
Вспомогательные методы диагностики позволяют выявить сопутствующие нарушения внутренних органов при синдроме Рубинштейна-Тейби. К таким методам относят ультразвуковые исследования (УЗИ почек и мочевыделительных путей, ЭхоКГ), электрокардиографию, магнитно-резонансную томографию и другие методики. Нередко у больных синдромом Рубинштейна-Тейби диагностируют врожденные пороки сердца (открытый Боталлов проток, дефект межжелудочковой перегородки), аритмии, аномалии почек (удвоение, гипоплазия) и мочевыделительных путей (атрезии на различных участках). Также при этом заболевании нарушается формирование мозолистого тела головного мозга, что определяется при помощи магнитно-резонансной томографии. У части больных синдромом Рубинштейна-Тейби отмечаются пороки развития легких и пищеварительной системы.
Специфического лечения данной патологии на сегодняшний момент не существует, все терапевтические мероприятия сводятся к облегчению симптомов, коррекции аномалий и пороков развития, угрожающих жизни больных. Назначаются ноотропные средства, препараты кальция и витамина Д, для уменьшения выраженности умственной отсталости рекомендуют специализированную психологическую помощь. В ряде случаев при синдроме Рубинштейна-Тейби применяют хирургические методы лечения – для устранения пороков сердца, аномалий развития прямой кишки и мочевыделительных путей, крипторхизма. Также может потребоваться лечение у офтальмолога, а при признаках развития злокачественного новообразования – консультация и лечение у онколога.
Прогноз и профилактика синдрома Рубинштейна-Тейби
Согласно мнению большинства специалистов, прогноз синдрома Рубинштейна-Тейби чаще всего неопределенный или неблагоприятный. Это связано с тем, что данное заболевание характеризуется значительным спектром разнообразных нарушений – от относительно безопасных для жизни (легкие скелетные аномалии, умственная неполноценность) до ярко выраженных, способных привести к смерти в раннем возрасте (тяжелые пороки развития сердца, легких, почек). Кроме того, значительно повышен риск развития онкологических патологий. Поэтому прогноз синдрома Рубинштейна-Тейби составляется строго индивидуально, исходя из конкретной клинической картины и общего состояния больного. Каких-либо специфических методов профилактики этого заболевания из-за его частого спонтанного развития на сегодняшний день не разработано.
МКБ-10
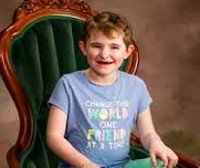
Синдром Ангельмана ( Синдром марионетки , Синдром Петрушки , Синдром счастливой куклы )
Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития. Проявляется интеллектуальным отставанием, слабой сформированностью речи, навыков сидения и ходьбы, хаотичными движениями, гиперактивностью, симптоматической эпилепсией, беспричинным весельем и смехом, сколиозом, своеобразной походкой. Пациенты имеют особую внешность: рот крупный, зубы расположены редко, подбородок выдается вперед. Диагноз устанавливается на основании клинических данных, результатов генетического анализа. Специфическое лечение отсутствует, проводится симптоматическая терапия, оказывается психологическая и педагогическая помощь.
Причины
Синдром Ангельмана – врожденное заболевание, в основе которого лежит генетический дефект. Патология развивается внутриутробно при делении хромосом. Нарушения в строении 15 хромосомы, переданной от матери, вызывают развитие болезни. Хромосома мутирует в результате делеции — выпадении генов, дупликации — появлении лишних генов, инверсии — обратном расположении генов, инсерции — изменении месторасположения генов, транслокации — присоединении участка одной хромосомы к другой.

- Дупликация хромосомы является причиной особой формы синдрома, несовместимой с жизнью. Если больные не умирают в младенчестве и достигают половой зрелости, детей они не имеют.
- Делеция хромосомы — причина тяжелой умственной отсталости. Больные дети не могут ходить и говорить, страдают от частых приступов эпилепсии, интенсивность которых имеет крайнюю степень выраженности.
- Мутация одного гена вызывает самую легкую форму синдрома, при которой больные дети могут себя обслуживать и общаться в коллективе. При этом они все равно отстают в развитии от своих сверстников.

Синдром Ангельмана возникает спонтанно. Любой ребенок может появиться на свет с данным недугом. Риск рождения значительно повышается в семьях, где у одного из родителей имеется такой же синдром или уже есть больной ребенок. У абсолютно здоровых родителей могут родиться больные дети. Мутация генов происходит под влиянием факторов, оказывающих тератогенное воздействие на плод:
- Вредные привычки беременной женщины,
- Всплеск эмоций, тяжелые душевные переживания, частые конфликты,
- Воспалительные заболевания органов женской репродуктивной системы,
- Длительный прием психотропных лекарств, наркотических веществ,
- Ионизирующее излучение.
Патогенез
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской хромосоме. Этот ген кодирует производство протеина Е6АР, который представляет собой ферментный компонент сложной реакции деградации белков. Е6АР участвует в процессе образования убиквитина – белка системы протеасом, стимулирующего протеолиз дефектных белковых молекул в нейронах головного мозга. В норме убиквитин маркирует ненужные (неактивные, нефункциональные) белки с целью инициации их уничтожения. Е6АР обеспечивает закрепление убиквитина на поверхности молекулы белка-мишени. Потом протеасомы расщепляют его на пептидные остатки и на аминокислоты. При синдроме Ангельмана убиквитин не закрепляется на дефектных белках, они скапливаются в нервной ткани мозга, нарушается процесс синаптической передачи. Формируются отклонения, задержки в психическом и моторном развитии.
Осложнения
Синдром Ангельмана – редкое малоизвестное заболевание. В связи с этим постановка диагноза и оказание медико-психолого-педагогической помощи зачастую проводятся несвоевременно, к 6-8 годам, что обуславливает низкую успешность коррекционных и лечебных мероприятий. Без физиотерапевтического лечения усугубляются нарушения опорно-двигательного аппарата – больные страдают от тяжелых форм сколиоза, самостоятельно передвигаются с трудом. Своеобразие внешности и поведения становится причиной ухудшения и без того затрудненной социальной адаптации. Все перечисленное влечет за собой утяжеление инвалидизации пациентов.
Диагностика
Диагностика синдром Ангельмана в младенческом возрасте вызывает определенные трудности у специалистов, поскольку заболевание не имеет специфической и явной симптоматики. Диагностические мероприятия делятся на пре- и постнатальные. Постнатальная диагностика основывается на характерных клинических признаках и внешних данных больного. Если симптоматика синдрома выражена незначительно, возникают проблемы с постановкой диагноза.
Первые клинические признаки патологии, на которые следует обратить внимание — гипотонус мышц, нарушающий процессы сосания и глотания. Мышечная слабость приводит к трудностям во время ходьбы. Больные дети ходят на негнущихся ногах, имеют особенное выражение лица, часто смеются, отстают в развитии общей моторики и речи от сверстников. У них выражен мелкий тремор и порывистые движения конечностей.

Специалистам необходимо собрать наследственный анамнез. Вероятность рождения больного ребенка у родителей, имеющих различные геномные или хромосомные нарушения, очень велика. В таких случаях генетический анализ проводят до рождения малыша.
Пренатальная диагностика заключается в проведении:
- УЗИ плода,
- Исследовании венозной крови беременной женщины на сывороточные маркеры хромосомных заболеваний,
- Биопсии ворсин хориона,
- Анализа околоплодной жидкости,
- Исследования плацентарной крови,
- Фетоскопии.
Во время генетического исследования находят дефект в 15 хромосоме и обнаруживают мутацию ответственного за заболевание гена. Генетическое исследование начинают с забора материала. Для этого у беременной женщины берут околоплодные воды или кровь и переходят непосредственно к анализу. Под микроскопом изучают ДНК, помеченную специальным красителем, и анализируют выявленные мутации в генах. Подобные диагностические методики дают точные результаты. С учетом наследственной предрасположенности и клинической картины ставят диагноз патологии. При наличии хромосомных отклонений у плода будущие родители должны принять осознанное решение.
Вспомогательными методами диагностики являются:
- Томографическое исследование, определяющее состояние и размеры головного мозга,
- Электроэнцефалография, показывающая работоспособность отдельных отделов мозга.
Для синдрома Ангельмана характерно отсутствие отклонений во время внутриутробного развития плода и врожденных аномалий у новорожденного. Результаты диагностических исследований часто остаются нормальными, а при проведении МРТ или КТ не обнаруживаются структурные изменения в головном мозге.
Окончательный диагноз ставят в возрасте 3-7 лет, когда четко видна динамика развития болезни с характерными клиническими проявлениями.
В статье расскажем всё о синдроме Ангельмана: определение, историю, причины, симптомы, приведём пример из практики с фото и видео. По каким признакам узнать и как лечить болезнь Ангельмана.
Отличительные признаки синдрома Ангельмана
Марионеточная походка, приступы смеха, радостное лицо.
Распространенность синдрома Ангельмана 1 на 10-30 тысяч новорожденных. Но большое количество случаев остаются не диагностированными, а наблюдаются у невролога с задержкой речевого развития, нарушениями поведения, эпилепсией. Мальчики и девочки болеют с одинаковой частотой. При изучении частоты встречаемости среди пациентов с умственной отсталостью выявлено 4,8% больных синдромом Ангельмана.
История
Заболевание названо по имени детского врача из Британии Гарри Ангельмана.

Harry Angelman
В 1965 году Эйнджелмен (вариант написания) описал трёх мальчиков из разных семей, назвал «Синдром счастливой марионетки» или «happy puppet syndrome», по их ангельскому счастливому внешнему виду и приступам беспричинного смеха, резким движениям в руках, дёрганной марионеточной походкой, тяжелой умственной отсталостью. Аналогия была проведена Эйнджелменом, глядя в музее Castelvecchio Вероны на картину «Мальчик – марионетка» с изображением смеющегося мальчика.
Чарльз Уильямс и Джейми Фриас в 1982 году описали ход болезни и предложили по этическим соображениям термин «синдром счастливой марионетки» заменить по имени описавшего педиатра в Синдром Ангельмана (СА).
Факты сегодняшней жизни
Среди популярных людей в настоящее время имеют в своей семье детей с Синдромом Ангельмана: Актер Колин Фаррелл, автор Иан Ранкин, профессиональный бейсболист Дэйв Хендерсон, профессиональный хоккеист Питер Мак Дафф.

Colin Farrell с сыном Henry
Этиологии патогенез

Хромосомы человека
Выявлено четыре генетических варианта появления синдрома Ангельмана:
- Мутация вновь возникшая – делеция в локусе 15 q11—q13 составляет большинство до 80 % всех случаев болезни.
- Диссомния по отцовской линии при потере передачи материнского локуса составляет 5% от всех случаев.
- Дефект центра импритинга до 5 %.
- Спонтанная мутация материнской копии вызывает отсутствие экспрессии родительской копии гена UBE3A в мозге. UBE3A кодирует деятельность фермента E6-AP убиквитин лигазы, которая выбирает один из четырех субстратов Е6-AP. Дефицит фермента E6-AP убиквитин лигазы, входящего в сложный процесс распада белков в гиппокампе – молекулярный механизм синдрома.
- У 7-9 % генетический синдром не идентифицируется.

Риск повторного рождения детей с синдромом Ангельмана
Тип генетической передачи определяет тяжесть клинических проявлений: при делеции делеция в локусе 15 q11—q13 (1 вариант) более грубые умственные и двигательные нарушения, резистентные к терапии эпилептические приступы.
При этом виде мутации происходит снижение ГАМК – рецепторов типа А.
Критерии диагноза синдрома Ангельмана
Симптомы, полезные в качестве поддерживающих критериев, но отклонение от них не исключает диагноза СА:
Нормальное течение перинатального периода; нормальная окружность головы при рождении; отсутствие выраженных пороков развития.
- Очевидная задержка развития, начиная с возраста 6- 12 месяцев жизни.
- Отсутствие прогрессирующей утраты приобретённых навыков.
- Нормальные результаты метаболических, гематологических и биохимических лабораторных тестов.
- Отсутствие структурных изменений в головном мозге по данным МРТ или КТ. Часто описывают умеренную кортикальную атрофию, проявление дисмиелинизации.
- Выраженная задержка психического развития.
- Речевые нарушения: отсутствие речи или скудный словарный запас (не более 6 слов).
- Возможно невербальное общение.
- Атаксия (это расстройство координации)
- Тремор конечностей
- Специфические особенности поведения: гиперактивность, стереотипии в виде размахивания руками; частый беспричинный смех.
Часто встречающиеся симптомы (у 80 %)
- Плоский затылок с канавкой.
- Высунутый язык. Отсюда и проблемы при сосании, жевании.
- Слюнотечение.
- Широкий рот с широкими редкими зубами.
- Прогнатия (это выступающая вперед нижняя челюсть).
- Косоглазие.
- Гипопигментация кожи, светлые волосы и глаза (только в случае делеции).
- Усиления сухожильных рефлексов с ног.
- Приподнятые плечи и полусогнутые в локтевых суставах руки при ходьбе.
- Плохая переносимость душных помещений.
- Нарушения сна.
- Повышенное внимание и притяжение к воде.
- В ответ на поднесённый к уху звучащий камертон больные широко улыбаются, смеются и наклоняются к камертону. Это может служить дополнительным диагностическим признаком при осмотре больных старше 1 года.
Клинические проявления синдрома Ангельмана
Внешний вид людей с синдромом Ангельмана .
Особенный черепно-лицевой и скелетный дисморфизм:
- Микроцефалия
- Гипоплазия средней части лица
- Глубоко посаженные глаза
- Выступающая вперед нижняя челюсть
- Протрузия языка (язык высунут и широкий)
- Заострённый подбородок
- Широкие межзубные промежутки
- Часто бледная кожа, светлые волосы, голубые глаза
- Укорочение сухожилий
- Сколиоз
Часты соматические и вегетативные расстройства : склонность к запорам, пищеводный рефлюкс, диффузная гипотония мышц, плохая переносимость жары.
При проведении неврологического осмотра выявляются:
Специфическая атаксическая походка: как марионетка – кукла на канатиках; «дерганная».
Резкие движения руками помогают удерживать тело в пространстве, балансировать.
Легкий тремор в покое.
Как распознать синдром Ангельмана
Частые симптомы синдрома Ангельмана :
Центральная нервная система
ЭЭГ
- высокоамплитудные пики и медленные волны с частотой 2- 3 Гц преимущественно в лобных отделах,
- высокоамплитудные медленные волны,
- пики в затылочных отделах, усиливающиеся при закрытых глазах,
- генерализованная высокоамплитудная медленная активность на протяжении почти всей записи 92%.
На МРТ и КТ картина неспецифическая:
- атрофия больших полушарий (33 %), расширение сильвиевых борозд, нейрональные гетеротопии, атрофия мозжечка, задержка миелинизации.
Кожа
Редкие симптомы
Течение и прогноз при Синдроме Ангельмана
- Умственная отсталость не прогрессирует, но остаётся очень тяжелой.
- Эпилептические припадки особенно частые и тяжелые в возрасте около 4 лет, а к 10-14 годам могут прекратиться.
- Смех вызван не положительными эмоциями, а поражением на уровне ствола мозга.
- У больных снижена потребность во сне, особенно в возрасте 2-6 лет.
- Несмотря на отсутствие речи, они могут общаться жестами.
- Развивается способность выполнять простые словесные инструкции.
- Некоторые могут пользоваться горшком к 4-5 годам.
- Каждый с СА не способен жить без постоянного ухода.
Лечение Синдрома Ангельмана
Медикаментозное лечение синдрома Ангельмана состоит:
- Противоэпилептической терапии;
- Коррекция поведенческих нарушений;
- Улучшения сна;
- Помощь в овладении навыками (двигательными, речевыми, по уходу);
- Адаптивная физическая культура.
Как общество относится к инвалидам, для чего необходимо адаптивное физическое развитие читайте в статье Паралимпиада в Сочи 2014.
Эпилептические приступы часто резистентны к терапии. С возрастом к 10 годам происходит урежение приступов.
В лечении эпилепсии при Синдроме Ангельмана используют:
Не используют в терапии СА в монотерапии из-за возможной аггравации приступов: фенитоин, карбамазепин, окскарбазепин, вигабатрин.
Но хороший эффект в комбинации
9. При кортикальном миоклонусе эффект показал пирацетам в высоких дозах 140 мг/кг/сут внутривенно капельно.
10. Кетогенная диета.
11. Препараты ноотропного действия (осторожно).
12. Седативные препараты для улучшения сна, с подбором доз.
Приведем клинический пример, послуживший поводом этой публикации.
Родители дали согласие на публикацию этого примера.
В нашем медицинском центре наблюдалось несколько пациентов с синдромом Ангельмана. Обращались с первичными жалобами на эпилептические приступы. После проведенных обследований выставлялся диагноз Синдром Ангельмана.
На приём в очередной раз обратились за выпиской для МСЭ родители с ребенком 6 лет 7 месяцев.
Сохраняются миоклонические судороги в момент засыпания, почти каждый вечер, от 0 до 10 раз подряд, стали менее интенсивные. Иногда мама чувствует миоклонии только своей рукой, положив руку на тело ребенка.
Сохраняется задержка психоречевого и моторного развития, отмечается легкая положительная динамика.
Самостоятельно ходит с 4 лет. Шаткая походка. Двигательная активность улучшилась. Танцует под музыку. Пытается сам есть ложкой.
Стал больше понимать, выполняет простые команды, стал хаотично возить ручкой по листу. Книжки листает, непродолжительно занимается с игрушками, смотрит мультфильмы.
Периодически раздражителен. Беспокойный сон (двигательное беспокойство во время сна, поверхностный).
Анамнез. Беременность на фоне хронической гипоксии плода. Роды 2, в 37 недель, экстренное кесарево сечение. Масса при рождении 3400, длина 54 см, по Апгар 7/8 баллов.
Формула развития: голову удерживает с 2 месяцев, сидит самостоятельно с 12 месяцев, ходит у опоры с 2,5 лет. В речи к 6,5 годам – говорит 3-5 слов с 3,6 лет.
С рождения часто срыгивал, расценивали как течение дисбактериоза.
НСГ – признаки гидроцефалии.
В возрасте от 1 до 4 месяцев отмечались пароксизмы вскидывания рук перед собой, и как бы покачивание в руках. После курса Кортексина и Церебролизина, Цераксона все купировалось.
Диагноз ДЦП выставлен в 1 год 6 месяцев. Проводили курсы реабилитации, включая медикаментозное лечение прозерином, кортексином; физиолечение (электрофорез).
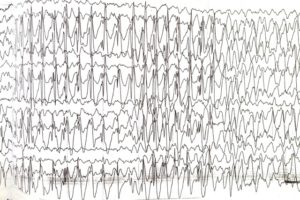
Эпиактивность при Синдроме Ангельмана
В 02.2012 году отмечался однократный эпизод, когда на фоне полного здоровья, после активного курса реабилитации, возникло состояние: активные миоклонии в плечевом поясе, кластерами; через 1 час появились падения назад с обмяканием, которые возникали после миоклоний. Между пароксизмами приходил в себя, то есть утрата контакта была при обмяканиях на несколько секунд. Пароксизмы продолжались по несколько секунд и длились до 1,5 суток, пока не ввели конвулекс. На фоне терапии конвулекса (35 мг/кг/сут) и дексазона пароксизмы стали уменьшаться. Приступы купированы через 3 суток.
В стационаре назначен дексаметазон №9, конвулекс по 75 мг * 2 раза в день длительно. Приступы были купированы, но отмечались побочные действия в виде интенсивного тремора.
С 05.2012 года конвулекс самостоятельно отменен. Приступы не отмечались.
Возобновились вздрагивания при засыпании с 07.2012 года.
Анализ на синдром Ангельмана сдан. Диагноз подтвержден в 06.2013 году.

Генетический анализ на Синдром Ангельмана
Заключение генетика: ish del (15)(q 11.2 q 11.2)(D15S11-, SNRPN-, D15S10-, GABR3-)
Отсутствуют 4 локуса хромосомы 15 в критической области синдромов Прадера – Вилли и Ангельмана (D15S11, SNRPN, D15S10, GABR3).
Переехал на постоянное место жительства из другого региона в 2012 году.
Состояние с положительной динамикой. Миоклонии стали реже, улучшилась походка, стал активнее, говорит несколько слогов.
С 06.2013 года был введен суксилеп. Динамика положительная.
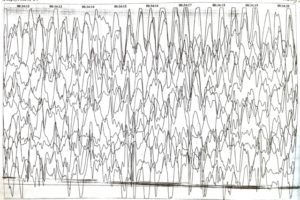
ЭЭГ синдром Ангельмана

ЭЭГ синдром Ангельмана
На осмотре невролога
Кожа и видимые слизистые чистые, бледные, волосы белесые.
Зев спокоен. Лимфоузлы не увеличены, отеков нет. В легких дыхание везикулярное, хрипов нет. Тоны сердца ясные ритмичные. ЧДД 20 в мин. ЧСС 84 уд в мин. Живот мягкий, безболезненный. Печень, селезенка не увеличены. Мочеиспускание свободное. Стул оформленный.

Обращает на себя внимание фенотип : маленький череп, гипоплазия средней части лица, макростомия (широкий рот), широкие межзубные промежутки, атаксия, задержка психо –речевого развития, мышечная гипотония, приступы смеха, сухожильная гиперрефлексия.
Неврологический статус
Состояние стабильное. Сознание ясное, реакция на осмотр не адекватная. Улыбчив, добродушен, легко идёт на контакт. Команды выполняет избирательно, не все понимает. Гиперактивен.
Череп микроцефальной формы (микробрахиоцефалия), окружность головы 45 см.
Глазные щели симметричны, объем движения глазных яблок полный, реакция зрачков на свет живая D=S. Лицо симметрично, носогубные складки не сглажены. Слух не нарушен. Глотает самостоятельно, не поперхивается. Язык по средней линии, высунут изо рта.
Сила мышц в конечностях 5 баллов, D=S. Тонус мышц снижен. Сухожильные рефлексы с рук и ног высокие, D=S. Патологические знаки + -.
Расстройства чувствительности нет.
Координационные пробы не понимает. Походка атаксическая, ходит на широко расставленных ногах.
Менингеальных знаков нет.
Итак, в приведенном примере мы увидели все характерные черты синдрома Ангельмана; проследили, как начинались и менялись эпилептические приступы и картина ЭЭГ; эффективность терапии. Встретив пациента с похожими симптомами, мы заподозрим и направим к генетикам на уточнения диагноза, выберем более верную тактику лечения.
Прогноз и профилактика
Выраженность симптомов синдрома Ангельмана может сильно различаться. Пациенты с легкими формами болезни имеют благоприятный прогноз: их речь становится более развернутой, улучшаются навыки самоконтроля при некоторых нарушениях двигательной сферы. При любой степени тяжести раннее начало и регулярное проведение медико-психологической помощи повышает качество жизни больных. Профилактика сводится к генетическому обследованию пар, в семьях которых есть ребенок с данным синдромом. Характер хромосомного дефекта (спорадический или наследственный) позволяет определить риск рождения второго больного ребенка.
Причины синдрома Рубинштейна-Тейби
Синдром Рубинштейна-Тейби обладает выраженной генетической гетерогенностью, однако различные типы этого заболевания не имеют каких-либо особенностей клинического течения. Наиболее часто причиной этой патологии выступают повреждения гена CREBBP, который расположен на 16-й хромосоме. Он кодирует так называемый CREB-связывающий белок, являющийся важным транскрипционным фактором, контролирующим экспрессию огромного количества генов. Появление в гене CREBBP различных изменений (точечных мутаций, транслокаций, микроделеций в 16-й хромосоме) приводит либо к образованию дефектного белка, неспособного выполнять свои функции, либо к полной блокировке его выделения. Это становится причиной развития синдрома Рубинштейна-Тейби.
В последние годы молекулярно-генетические исследования данной патологии подтвердили, что в ее развитии играют роль и мутации гена EP300, который располагается на 22-й хромосоме. Продуктом экспрессии данного гена является протеин p300, который, как и CREB-связывающий белок, относится к группе транскрипционных факторов. По данным исследований, идентифицировать характер генетического дефекта при синдроме Рубинштейна-Тейби удается в 55-60% случаев, примерно у половины пациентов обнаруживаются мутации гена CREBBP, еще у 40-45% выявляются делеции и другие хромосомные перестройки, затрагивающую 16-ю хромосому, и лишь у 3% больных наблюдаются мутации гена EP300. Все это указывает на возможность участия других генов в развитии этого заболевания.
Дефект белка-фактора транскрипции или его малое выделение (по причине гаплонедостаточности) приводит к многочисленным нарушениям, обусловленным недостаточной стимуляцией ряда других генов. Это можно заметить по характерной клинической картине синдрома Рубинштейна-Тейби. Оба транскрипционных фактора влияют на активность образования новых связей между нейронами, формирование долговременной памяти, стимуляцию иммунного ответа, развитие репродуктивной системы. Недостаточность этих белков ведет к целому каскаду патологических процессов в организме, что и составляет клинику синдрома Рубинштейна-Тейби. Кроме того, вышеуказанные транскрипционные факторы также влияют на активность антионкогенов, поэтому при их дефекте значительно возрастает риск развития злокачественных новообразований.
Синдром Рубинштейна-Тейби
Синдром Рубинштейна-Тейби – генетически гетерогенное (по последним данным) наследственное заболевание, характеризующееся поражением центральной нервной системы, деформациями костей скелета и рядом других пороков развития. Симптомами этого состояния являются прогрессирующая умственная отсталость, низкий рост, расширение фаланг пальцев, полидактилия на ногах, разнообразные нарушения со стороны внутренних органов. Диагностика синдрома Рубинштейна-Тейби производится на основании данных настоящего статуса пациента, молекулярно-генетических анализов и других исследований. Специфического лечения данной патологии не существует, применяют симптоматическую терапию в зависимости от типа пороков и нарушений.
Общие сведения
Синдром Рубинштейна-Тейби – генетическое заболевание, которое сопровождается нарушениями интеллектуального и физического развития, а также разнообразными пороками скелета и внутренних органов. Впервые данная патология была выявлена в 1963 году и описана американскими педиатрами Дж. Рубинштейном и Г. Тейби – сами исследователи назвали заболевание «синдромом широкого первого пальца кистей и стоп, специфического лица и умственной отсталости». Дальнейшие исследования в области генетики подтвердили, что синдром Рубинштейна-Тейби является аутосомно-доминантной патологией, но подавляющее большинство случаев возникает вследствие спонтанных мутаций различного типа. Встречаемость составляет порядка 1:100 000-125 000, мальчики и девочки поражаются примерно с одинаковой частотой. Особенностью синдрома Рубинштейна-Тейби является повышенный риск развития различных онкологических заболеваний – главным образом, некоторых типов лейкозов, опухолей мозговых оболочек и нервных тканей.
Симптомы синдрома Рубинштейна-Тейби
Симптоматика синдрома Рубинштейна-Тейби характеризуется значительным разнообразием у разных больных, что отражается на тяжести течения заболевания и его прогнозе. Обычно уже при рождении можно обнаружить некоторые признаки этой патологии – деформации лица и черепа (микро- или брахицефалия, расширение переносицы, эпикант, клювовидный нос), высокое арковидное нёбо, изменение формы и положения ушных раковин, расширенные фаланги пальцев, особенно первых. На ногах у больных синдромом Рубинштейна-Тейби нередко выявляется полидактилия. Иногда уже при рождении определяются признаки пороков развития внутренних органов – бледность или цианоз (при поражении легких или сердца), длительная желтуха новорожденных, крипторхизм.
При дальнейшем течении синдрома Рубинштейна-Тейби отмечается прогрессирующее отставание ребенка в физическом и умственном развитии от здоровых сверстников, затрудненное развитие речи и моторных навыков. Также наблюдается косоглазие, миопия. Выражение лица больных характеризуется гримасой, напоминающей улыбку, возникает гирсутизм, примерно у половины пациентов определяется наличие красного невуса в области лба, затылка или шеи. В старшем возрасте у больных синдромом Рубинштейна-Тейби выявляются искривления позвоночного столба различного характера и низкий рост (не более 150-160 сантиметров). Могут определяться деформации грудной клетки и (реже) другие скелетные аномалии, у мальчиков в большинстве случаев отмечается крипторхизм.
Постоянным признаком синдрома Рубинштейна-Тейби является умственная отсталость. Как правило, диагностируется ЗПР, олигофрения, значительная задержка речевого развития по сравнению со здоровыми сверстниками. Отмечаются нарушения концентрации внимания, больные легко отвлекаются на посторонние раздражители при выполнении какого-либо задания, возможны резкие перепады настроения. При этом лица с синдромом Рубинштейна-Тейби хорошо идут на контакт, легко социализируются. Среди других неврологических симптомов заболевания нередко обнаруживается плохая координация движений, изредка наблюдаются судорожные приступы.
Синдром Рубинштейна-Тейби может также проявляться различными нарушениями со стороны внутренних органов – головного мозга, сердца, почек и мочевыделительных путей, терминальных отделов пищеварительной системы. Кроме того, у таких больных значительно повышается риск возникновения онкологических заболеваний, в основном развивающихся в раннем возрасте. К ним относят различные формы лейкозов, меланому, некоторые типы лимфом. Поэтому до пубертатного периода больные синдромом Рубинштейна-Тейби должны периодически проходить обследования у онколога для ранней диагностики злокачественных новообразований.
Общие сведения
Синдром назван по фамилии британского педиатра Г. Ангельмана. В 1965 году он первым описал симптомы заболевания и назвал его «синдромом счастливой марионетки», поскольку пациенты напоминали ему героя картины «Мальчик-марионетка». В те годы методы генетических исследований были еще не разработаны, установить причину патологии было невозможно. В 1987 году исследователи определили этиологию болезни и переименовали ее в синдром Ангельмана. Сейчас этот термин является официальным, но можно встретить синонимичные названия – «синдром марионетки», «синдром Петрушки», «синдром счастливой куклы». Распространенность составляет 1 случай на 10-20 тысяч новорожденных. Заболевание выявляется после первого года жизни (иногда – к 3-7 годам), чаще болеют мальчики.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
Всё о синдроме Ангельмана
Симптомы
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно нарастает задержка развития, ранее освоенные навыки сохраняются, но приобретение новых происходит медленно. Дети умеют сидеть, ползать, брать предметы и перекладывать их из руки в руку, поддерживать визуальный контакт, гулить и лепетать. Ходьба дается с трудом, нарушено чувство равновесия, наблюдаются частые падения, ушибы о мебель. Выявляется тремор и хаотичные движения конечностями, особенно руками. Речевые расстройства представлены как задержками, так и полным отсутствием экспрессивной речи. Дети либо совсем не говорят, либо используют лепет, простые слоги и слова общим объемом не более 10 единиц. Сохраняется понимание обращенной речи, стремление к общению, использование невербальных средств коммуникации: жестов, мимики, опосредованных знаков.
Основное поведенческое нарушение – гиперактивность. Дети часто веселятся и смеются без объективной причины, двигательно расторможены, неусидчивы, нецеленаправленны. У многих возникает патологическая привязанность к определенной игрушке или предмету быта, при появлении которого настроение сразу повышается, капризность и плач сменяются смехом. Концентрация внимания снижена, переключаемость быстрая и ненаправленная. Имеются трудности обучения, стойкое снижение интеллектуальных функций. Легко закрепляются стереотипии: раскачивание тела, размахивание руками. У 80% пациентов отмечается микроцефалия, недостаточный объем черепной коробки, эпилептическая активность мозга. Редко наблюдается снижение контроля движений языка, которое проявляется трудностями сосания груди или соски, последующим недостатком массы тела.
Характерные особенности внешности детей – косоглазие, сколиотическое искривление позвоночника, увеличение зубов и губ, разряжение зубного ряда, уплощение затылка, выступание вперед подбородка. Язык часто высунут, рот приоткрыт в улыбке. Развивается мышечная дистония, выраженность рефлексов сухожилий повышается, формируя специфичность моторики: пациенты ходят на прямых несгибающихся ногах, плечи приподнимают, руки сгибают в локтях. Своеобразный симптом – тяга к воде. Большинство детей чувствуют себя спокойнее в водной среде, им нравится плескаться в ванной, играть с корабликами в тазу.
Видео: о детях с синдромом Ангельмана
Прогноз
Прогноз патологии определяется степенью поражения 15 хромосомы. Больные с незначительными изменениями могут сами себя обслуживать и нормально вести себя в обществе. Дети с выраженным генетическим дефектом не разговаривают и практически не ходят. Тяжелее всего протекает недуг у детей, в чьей хромосоме отсутствуют участки генов. Они обычно не могут самостоятельно ходить и говорить. Все остальные формы патологии поддаются коррекции. В любом случае стать полноценными членами общества такие больные не смогут. Они понимают речь, но не вступают в диалог. Их с детства обучают языку жестов. Вербальное общение им недоступно.
Больные с синдромом Ангельмана требуют особого внимания и заботы. Родные и близкие люди должны создать для больного атмосферу понимания, любви и дружелюбия. Больным детям требуется помощь в адаптации к жизни в обществе. Им показан специальный режим питания и сна, тщательный уход, обучение элементарным навыкам самообслуживания. Существуют специализированные интернаты для таких детей, предполагающие специальный курс адаптации к социуму и обучения. Больных учат языку жестов и проводят занятия по особым программам, направленным на развитие мелкой и общей моторики.

С возрастом гиперактивность и нарушения сна проходят самостоятельно. Ели лечение начато рано и проводится грамотно, прогноз патологии улучшается. На продолжительность жизни заболевание не влияет.
При грамотном лечении и внимательном отношении можно значительно облегчить жизнь людям с синдромом Ангельмана, но они никогда не смогут жить самостоятельно. Эта та категория лиц, которая будет постоянно нуждаться в опеке и заботе.
Лечение
Синдром Ангельмана — неизлечимая генетическая аномалия. В настоящее время ученые-медики активно разрабатывают эффективные терапевтические методики. Если синдром был диагностирован во время внутриутробного развития, специалисты рекомендуют прервать беременность. Новорожденному больному ребенку необходим тщательный уход, забота и высококвалифицированная терапия.

Симптоматическое лечение помогает облегчить состояние больных с синдромом марионетки. Им назначают лекарственные препараты и немедикаментозные процедуры:
Диагностика
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и генетики. Родители предъявляют жалобы на отсутствие речи, двигательные стереотипии, трудности формирования ходьбы и других двигательных навыков, гиперактивность. Проводится дифференциальная диагностика, в ходе которой должны быть исключены более распространенные заболевания, такие как умственная отсталость, расстройства аутистического спектра, деменции, мутизм, несимптоматические формы эпилепсии. Комплексное исследование включает следующие процедуры:
- Общий осмотр. На наличие синдрома часто указывают специфические черты внешности пациентов: высунутый язык, слюнотечение, крупный рот, широкие редкие зубы, выступающая вперед нижняя челюсть, плоская форма затылка. Характерен светлый оттенок кожи, глаз и волос. Походка детей напоминает движения куклы-марионетки из-за усиления сухожильных рефлексов и снижения мышечного тонуса.
- Осмотр психиатром. В 100% случаев синдрома Ангельмана диагностируется выраженная задержка в развитии психики, отсутствие самостоятельной речи или очень скудный словарный запас. Коммуникация осуществляется с помощью мимики, жестов, рисунков. В поведении отмечается гиперактивность, стереотипные движения руками, беспричинный смех.
- Неврологическое обследование. У всех пациентов определяется атаксия и тремор конечностей. 80% больных имеют постнатальную микроцефалию – окружность головы новорожденного меньше 32 см, к 12 месяцам – около 42 см. По данным ЭЭГ выявляется симптоматическая эпилепсия (высокоамплитудные разряды медленных комплексных волн), клинически возможны судорожные припадки. У некоторых детей имеется косоглазие, диффузное снижение мышечного тонуса, усиление сухожильных рефлексов, гиперкинезы.
- Генетическое исследование. Лабораторная диагностика нацелена на выявление мутаций и делеций в гене UBE3A. Последовательно проводится комплекс процедур, включающий флуоресцентную гибридизацию in situ, анализ мутации центра запечатления, метилирование ДНК СА/ПВС региона, диагностику делеции методом микроматричного анализа, поиск мутационных изменений в локусе UBE3A.
Профилактика
Предупредить рождение ребенка с синдромом Ангельмана невозможно, поскольку мутация генов происходит спонтанно. Объяснить подобный процесс не под силу даже самым квалифицированным генетикам.
Профилактические мероприятия направлены на тщательную подготовку к беременности и рождению ребенка. Все беременные женщины должны пройти в назначенный срок УЗИ плода и сдать кровь на сывороточные маркеры хромосомных патологий. В случае положительного результата медико-генетического исследования родители должны решить, смогут ли они воспитывать больного ребенка.
Медико-генетическое консультирование требуется парам с отягощенной наследственностью. Если данное заболевание не было зафиксировано у близких и родных, достаточно соблюдать основные принципы ведения здорового образа жизни во время беременности.
Синдром Ангельмана – тяжелая и неизлечимая болезнь, лишающая человека нормальной жизни. Больные дети нуждаются в особой заботе и внимании. Лечение направлено на снижении интенсивности симптомов патологии. Современные ученые стараются найти действенные способы борьбы с хромосомными аномалиями, жертвами которых становятся новорожденные дети.
Читайте также: