
Alagille syndrome что это
Обновлено: 04.07.2024
Синдром Алажилля – редко встречающееся генетически детерминированное заболевание с аутосомно-доминантным типом наследования, характеризующееся хроническим внутрипеченочным холестазом вследствие аномалии развития билиарного дерева в сочетании с другими врожденными пороками. Большие трудности представляет диагностика этого заболевания у детей раннего возраста, когда необходимо определить причину холестаза и тактику дальнейшего ведения ребенка.
Цель. Анализ особенностей клинических проявлений и лабораторных показателей у детей с синдромом Алажилля.
Характеристика детей и методы исследования. Проведен ретроспективный анализ клинико-диагностических проявлений синдрома Алажилля у 21 ребенка (10 мальчиков и 11 девочек) в возрасте от 1 мес до 14 лет 5 мес (средний возраст 5±1 год) – сплошное исследование. Изучались анамнез жизни и болезни пациентов с оценкой данных течения беременности у матерей, клинические проявления дебюта заболеваний, результаты клинико-диагностических исследований, в том числе в дебюте болезни и проведенные по месту жительства, а также при первой госпитализации в клинику. Всем детям осуществлялись ультразвуковое исследование органов брюшной полости, биохимическое исследование крови.
Результаты. Анализ клинико-диагностических критериев детей с синдромом Алажилля показал, что значимыми в диагностике являются угроза прерывания беременности у матерей, внутриутробная гипотрофия плода, затянувшаяся в неонатальном периоде желтуха, которая может сохраняться и в более старшем возрасте и сопровождаться гепато-/гепатоспленомегалией (чаще встречаются в возрасте старше 3 мес), гипо-/ахоличным стулом, который после 3 мес жизни наблюдается значительно реже, коагулопатией, более характерной для детей первых 3 мес жизни, а также изменениями биохимических показателей крови: характерно превышение верхней границы нормы для цитолитической активности в 2–5 раз, гипербилирубинемии (с преобладанием прямой фракции билирубина) – в 3–7 раз, уровня гамма-глютамилтранспептидазы – в 2–7 раз.
Заключение. Синдром Алажилля необходимо диагностировать максимально рано для определения дальнейшей тактики лечения. Тщательный сбор анамнеза, анализ клинико-диагностических проявлений заболевания, особенности изменений биохимических анализов крови позволяют провести диагностику этого заболевания.
Ключевые слова
Об авторах
ОСП «Научно-исследовательский клинический институт педиатрии им. академика Ю.Е. Вельтищева» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава РоссииРоссия
Волынец Галина Васильевна – д.м.н., гл. науч. сотр. отдела гастроэнтерологии Научно-исследовательского клинического института педиатрии им. академика Ю.Е. Вельтищева
125412 Москва, ул. Талдомская, д. 2.
ОСП «Научно-исследовательский клинический институт педиатрии им. академика Ю.Е. Вельтищева» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава РоссииРоссия
Никитин Артем Вячеславович – к.м.н., вед. науч. сотр. отдела гастроэнтерологии Научно-исследовательского клинического института педиатрии им. академика Ю.Е. Вельтищева, асс. кафедры гастроэнтерологии РНИМУ им. Н.И. Пирогова
125412 Москва, ул. Талдомская, д. 2.
ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава РоссииРоссия
Скворцова Тамара Андреевна – к.м.н., доц. кафедры гастроэнтерологии ФДПО РНИМУ им. Н.И. Пирогова, зав. отделением гастроэнтерологии Морозовской детской городской клинической больницы, гл. внештатный детский специалист-гастроэнтеролог Департамента здравоохранения города Москвы,
119049 Москва, 4-й Добрынинский пер., д. 1/9
Список литературы
1. Kamath B.M., Loomes K.M., Piccoli D.A. Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr 2010; 50(6): 580–586. DOI: 10.1097/MPG.0b013e3181d98ea8
2. Lykavieris P., Hadchouel M., Chardot C., Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut 2001; 49(3): 431–435. DOI: 10.1136/gut.49.3.431
3. Li L., Krantz I.D., Deng Y., Genin A., Banta A.B., Collins C.C. et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997; 16 (3): 243–251. DOI: 10.1038/ng0797-243
4. Oda T., Elkahloun A.G., Pike B.L., Okajima K., Krantz I.D., Genin A. et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 1997; 16(3): 235– 242. DOI: 10.1038/ng0797-235
5. McDaniell R., Warthen D.M., Sanchez-Lara P.A., Pai A., Krantz I.D., Piccoli D.A. et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet 2006; 79(1): 169–173. DOI: 10.1086/505332
6. Hofmann J.J., Zovein A.C., Koh H., Radtke F., Weinmaster G., Iruela-Arispe M.L. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development 2010; 137(23): 4061–4072. DOI: 10.1242/dev.052118
7. Turnpenny P.D., Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 2012; 20(3): 251–257. DOI: 10.1038/ejhg.2011.181
8. Mitchell E., Gilbert M., Loomes K.M. Alagille Syndrome. Clin Liver Dis 2018; 22(4): 625–641. DOI: 10.1016/j.cld.2018.06.001
9. Tsai E.A., Gilbert M.A., Grochowski C.M., Underkoffler L.A., Meng H., Zhang X. et al. THBS2 Is a Candidate Modifier of Liver Disease Severity in Alagille Syndrome. Cell Mol Gastroenterol Hepatol 2016; 2(5): 663–675.e2. DOI: 10.1016/j.jcmgh.2016.05.013
10. Hofmann J.J., Briot A., Enciso J., Zovein A.C., Ren S., Zhang Z.W. et al. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 2012; 139(23): 4449– 4460. DOI: 10.1242/dev.084871
11. Vajro P., Ferrante L., Paolella G. Alagille syndrome: an overview. Clin Res Hepatol Gastroenterol 2012; 36(3): 275–277. DOI: 10.1016/j.clinre.2012.03.019
12. De Bruyne R., Van Biervliet S., Vande Velde S., Van Winckel M. Clinical practice: neonatal cholestasis. Eur J Pediatr 2011; 170(3): 279–284. DOI: 10.1007/s00431-010-1363-8
13. Kamath B.M., Schwarz K.B., Hadzić N. Alagille syndrome and liver transplantation. J Pediatr Gastroenterol Nutr 2010;50 (1): 11–5. DOI: 10.1097/MPG.0b013e3181c1601f
14. Kamath B.M., Loomes K.M., Oakey R.J., Emerick K.E., Conversano T., Spinner N.B. et al. Facial features in Alagille syndrome: specific or cholestasis facies? Am J Med Genet 2002; 112(2): 163–170. DOI: 10.1002/ajmg.10579
15. Cho H.L., Kim J.S., Paeng S.S., Lee S.H. Butterfly vertebra with lumbar intervertebral disc herniation. J Neurosurg Spine 2011; 15(5): 567–570. DOI: 10.3171/2011.6.SPINE1178
16. Subramaniam P., Knisely A., Portmann B., Qureshi S.A., Aclimandos W.A., Karani J.B. et al. Diagnosis of Alagille syndrome – 25 years of experience at King’s College Hospital. J Pediatr Gastroenterol Nutr 2011; 52(1): 84–89. DOI: 10.1097/MPG.0b013e3181f1572d
17. Mainwaring R.D., Sheikh A.Y., Punn R., Reddy V.M., Hanley F.L. Surgical outcomes for patients with pulmonary atresia/major aortopulmonary collaterals and Alagille syndrome. Eur J Cardiothorac Surg 2012; 42(2): 235–240; discussion 240–241. DOI: 10.1093/ejcts/ezr310
19. Spinner N.B., Leonard L.D., Krantz I.D. Alagille Syndrome. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., Amemiya A., editors. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. PMID: 20301450.
20. Волынец Г.В., Хавкин А.И., Никитин А.В., Скворцова Т.А. Дифференциальная диагностика и принципы терапии врожденных холестатических болезней у детей раннего возраста. Монография. М.: Прима Принт, 2018; 160. [Volynets G.V., Khavkin A.I., Nikitin A.V., Skvortsova T.A. Differential diagnosis and treatment principles of congenital cholestatic diseases in young children. Monography. Moscow: Prima Print, 2018; 160. (in Russ.)]
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Текст научной работы на тему «Синдром Алажилля у девочки с синдромом Шерешевского-Тернера»
СИНДРОМ АЛАЖИЛЛЯ У ДЕВОЧКИ С СИНДРОМОМ ШЕРЕШЕВСКОГО—ТЕРНЕРА
Л. Я. Климов1, М. В. Стоян1, Т. М. Вдовина3, В. А. Курьянинова1, Л. В. Погорелова1, Р. А. Атанесян1, С. В. Долбня1, О. К. Кулешова1, О. И. Еремеева2, Т. А. Углова2, Т. И. Кадурина4 1ГБОУ ВПО Ставропольская государственная медицинская академия, 2Детская городская клиническая больница имени Г. К. Филиппского, 3Краевой клинический клинико-диагностический центр, Ставрополь, 4 ГБОУ ВПО Северо-Западный государственный медицинский университет им. И. И. Мечникова,
ALAGILLE SYNDROME OF THE GIRL WITH TURNER SYNDROME
L. Ya. Klimov1, M. V. Stoyan1, T. M. Vdovina3, V. A. Kuryaninova1, L. V. Pogorelova1, R. A. Atanesyan1, S. V. Dolbnya1, O. K. Kuleshova1, O. I. Eremeyeva2, T. A. Uglova2,
© Коллектив авторов, 2011 г.
В статье впервые в литературе представлено наблюдение аутосомно-доминантно наследуемого синдрома Алажилля в сочетании с мозаичным вариантом синдрома Шерешевского—Тернера у девочки 7 лет. Диагностировано столь редкое сочетание благодаря проведению биопсии печени у ребенка с синдромом внутри-печеночного холестаза и цитогенетического обследования в связи с отставанием в росте и множественными дизморфиями.
Ключевые слова: синдром Алажилля, внутрипеченочный холестаз, синдром Шерешевского—Тернера, мозаицизм.
Keywords: Alagille syndrome, intrahepatic cholestasis, Turner syndrome, mosaicism.
Введение. Синдром Алажилля (син. — артерио-печеночная дисплазия, синдром Алажилля— Ватсона) — генетическое заболевание с аутосом-но-доминантным типом наследования, которое характеризуется недостаточностью внутрипече-ночных желчных протоков и развитием синдрома внутрипеченочного холестаза, описанное Даниэлем Алажиллем в 1975 г. 2. У 70% больных синдром обусловлен мутациями гена JAG1, расположенного в сегменте 20р12, а у 7% находят де-леции или транслокации, затрагивающие тот же сегмент 5.
Синдром Алажилля проявляется внутрипе-ченочным холестазом, особенностями фенотипа, аномалиями позвоночника, почек, органа зрения, сердечно-сосудистой системы, задержкой роста и неврологическими нарушениями. Распростра-
ненность среди живорожденных детей составляет 1 : 70 000-1 : 100 000 4.
Синдром внутрипеченочного холестаза появляется в периоде новорожденности, чаще всего в течение первых 3 месяцев жизни. Клинически выявляются: желтуха с зеленоватым оттенком, увеличение печени, преимущественно за счет левой доли, спленомегалия при отсутствии портальной гипертензии, ахоличный стул и темный цвет мочи. К 4-6 месяцам жизни, как правило, наблюдается снижение уровня билирубина, нормализация цвета мочи и стула, однако появляется постоянный кожный зуд [5, 6].
У ряда больных встречаются ксантомы, обусловленные гиперхолестеринемией, которые, как правило, расположены на тыльной поверхности суставов пальцев, ладонной поверхности кистей
рук, задней стороне шеи, у основания волос, в радиальных складках ануса, в подколенных ямках, а иногда и в паховой области. Характерны особенности строения лицевого черепа: выпуклый лоб, энофтальм, умеренный гипертелоризм, прямой нос с плоским кончиком, уменьшенный, заостренный, приподнятый и направленный вперед подбородок, низко посаженные, а в ряде случаев — деформированные ушные раковины. Симптомами скелетных поражений являются аномалии дуг позвонков, создающие картину spina bifida, остеопороз, задержка костного возраста, укорочение дистальных фаланг, короткая локтевая кость.
Со стороны мочевыделительной системы описаны нарушение концентрационной функции, нефролитиаз, гипоплазия, дистопия, поликистоз почек. Среди аномалий сердечно-сосудистой системы в 85% случаев встречается гемодинами-чески незначимый стеноз или гипоплазия легочной артерии, гипертрофия правых отделов сердца, тетрада Фалло. Поражение органа зрения проявляется задним эмбриотоксоном (помутнение роговицы — Прим. ред.), различными пигментными изменениями и эктопией зрачка, страбизмом. По данным литературы, характерный фенотип встречается у 95% больных с синдромом Алажилля, при этом — хронический холестаз и гиперхолестеринемия — у 96%, патология органа зрения — у 83%, внутриутробная гипотрофия — у 70%, аномалии позвоночника — у 59%, а патология почек — у 57% [3, 5, 6, 10, 11]. У части больных отмечены гипогонадизм, задержка умственного развития, неврологические нарушения по типу периферической нейропатии [2-4, 6].
При морфологическом исследовании печени определяется уменьшение количества портальных желчных протоков, снижение отношения внутрипеченочных желчных протоков к портальным трактам менее 0,9. У новорожденных могут выявляться гигантоклеточная трансформация и холестаз. Выявляют задержку желчи в гепа-тоцитах на уровне комплекса Гольджи и необычно большое количество межклеточной желчи. В ряде случаев описаны признаки перипорталь-ного фиброза [3, 4, 6, 10].
Существенная задержка роста, диагностируемая у большинства пациентов, по мнению ряда исследователей, обусловлена нечувствительностью рецепторов к соматотропному гормону [12], однако это мнение не общепризнанно.
Прогноз для жизни больных в основном зависит от выраженности поражения печени: интенсивности и длительности холестаза, наличия печеночной недостаточности, портальной гипер-тензии и возникновения внутрипеченочных кро-
воизлияний. Неблагоприятным фактором, резко снижающим продолжительность жизни, является наличие врожденного порока сердца, особенно синего типа. В среднем 75% больных доживают до 20 лет [3].
Лечение синдрома Алажилля зависит от наличия и выраженности сопутствующих клинических проявлений. Трансплантация печени показана пациентам с печеночной недостаточностью, сильным зудом, не поддающимся медикаментозной коррекции и выраженной задержкой роста.
В связи с тем, что мы не встретили в литературе описания сочетания синдрома Алажилля с синдромом Шерешевского—Тернера, приводим собственное наблюдение.
Больная Ч., 7 лет, поступила в гастроэнтерологическое отделение ДГКБ имени Г. К. Филип-пского г. Ставрополя с жалобами на желтушное окрашивание кожи и склер, постоянный сильный кожный зуд, значительную задержку физического развития, периодические боли в животе, беспокойный сон.
содержания гамма-глутамилтрансферазы (ГГТ) до 124 Ед/л (норма 7-15 Ед/л), сидеропения — 1,7 мкмоль/л. В лечении использованы урсосан, эссенциале, фенобарбитал. На фоне терапии отмечалась слабо выраженная положительная динамика. Неоднократно исследовали маркерный спектр гепатитов, данных за вирусный гепатит не получено. При амбулаторном исследовании определялась гипербилирубинемия до 140 мкмоль/л (за счет повышения как прямой, так и непрямой фракций), нарастание уровня трансаминаз в пределах 2 норм, холестерина, ГГТ, диспротеинемия. УЗИ органов брюшной полости выявило признаки гепатомегалии и перегиба желчного пузыря. МРТ, выполненная в возрасте 3 лет, позволила исключить атрезию желчевыводящих путей.
С рождения у девочки наблюдалась выраженная задержка физического развития. Так, в возрасте 3 лет ее рост составил 81,5 см (-3,47 S), масса тела — 9,5 кг (-2,88 8); в 4 года соответственно — 87 см (-3,24 S) и 9,7 кг (-3,78 S); в 5 лет — 91 см (-3,51 S) и 11 кг (-2,81 S), в 6 лет — 93 см (-3,96 S) и 13 кг (-1,21 S).
i Не можете найти то, что вам нужно? Попробуйте сервис подбора литературы.В 3 года ребенок консультирован генетиком, проведено цитогенетическое обследование, ка-риотип — 46,ХХ (анализ более 11 метафазных пластин). Костный возраст отставал от календарного на 3 года. В связи с выраженным отставанием в физическом развитии и особенностями фенотипа был заподозрен синдром Рассела— Сильвера с сопутствующим нарушением пигментного обмена (синдром Ротора или синдром Дабина—Джонсона).
Длительное течение заболевания, стойкая ги-пербилирубинемия, повышенная активность трансаминаз, несмотря на лечение, послужили основанием для госпитализации в гастроэнтерологическое отделение РДКБ г. Москвы. Повторное цитогенетическое исследование с анализом 30 метафазных пластин выявило в 8 (24%) карио-тип 45,Х, а в 22 (76%) — 46,ХХ, что соответствует мозаичной форме синдрома Шерешевского— Тернера.
На УЗИ органов брюшной полости выявлены изменения печени с локальным участком гепато-за (VII сегмент), холангит, диффузные изменения поджелудочной железы. Результаты элас-тометрии соответствуют стадии фиброза F1 по METAVIR. Исследование биоптата печени выявило дискомплектацию балок, зернистую дистрофию цитоплазмы гепатоцитов, наличие единичных двуядерных гепатоцитов; щелевидные синусоиды, звездчатые ретикулоэндотелиоциты в состоянии мобилизации. Портальные тракты распределялись неравномерно, были частично
сближены и расширены за счет неравномерно выраженного фиброза и скудной смешанно-клеточной инфильтрации, не выходящей за терминальные пластинки. Желчные протоки достоверно не визуализировались, центральные вены распределялись неравномерно, были расширены, но оптически свободны, в некоторых из них отмечено утолщение стенок.
Рентгенография грудного отдела позвоночника выявила расщепление задней дужки ThVII тела позвонка с диастазом в 1 мм по типу spina bifida posterior и особое строение тел позвонков ThVII, ThVI по типу «бабочки».
Консультация окулиста: выявлено легкое венозное полнокровие и извитость сосудов глазного дна.
На основании результатов пункционной биопсии печени, рентгенографии позвоночника и данных повторного цитогенетического исследования установлен диагноз: Синдром атрезии внут-рипеченочных желчных протоков (синдром Алажилля), мозаичный вариант (45,Х/46,ХХ) синдрома Шерешевского—Тернера. Рекомендована трансплантация печени в плановом порядке при наличии донора. После выписки ребенок постоянно получал урсосан в дозе 250 мг/сут, однако существенная положительная динамика в состоянии отсутствовала.
При осмотре общее состояние ребенка средней степени тяжести, обусловленное сочетанной патологией. Рост — 101 см (-3,528, 3-я перцен-тиль), масса тела — 14 кг (-1,858, 3-я перцен-тиль), окружность головы — 48 см (3-я перцен-тиль), окружность грудной клетки — 52 см (3-я перцентиль), окружность живота — 52,5 см. Дефицит массы тела — 14,8%. Физическое развитие низкое, гармоничное (рис. 1).
В сознании, в контакт практически не вступает, негативна, плаксива, самочувствие нарушено. Телосложение астеническое, питание понижено. Фенотипические особенности: голова псевдогид-роцефальной формы, лицо — треугольное, лоб высокий, глазной гипертелоризм, энофтальм, высокая спинка носа с плоским кончиком, короткий фильтр, низко расположенные ушные раковины, дизотия, гипоплазия мочек ушных раковин, редкие и мягкие волосы, редкие брови, короткая шея с крыловидными складками, гипертелоризм сосков, увеличенный живот, вялая осанка, укорочение туловища, клинодактилия V пальцев кистей, короткие дистальные фаланги, «барабанные палочки», «часовые стекла», вальгусная деформация нижних конечностей (рис. 1, 2).
Кожные покровы и склеры иктеричны, сухие на ощупь, с множественными экскориациями и гиперкератозом ладоней. На IV пальце левой
Рис. 1. Общий вид девочки (описание в тексте)
кисти гиперпигментированное пятно диаметром около 1,5 см, на остальных пальцах — более мелкие гипер- и депигментированные пятна (рис. 3). Венозная сеть на лбу и передней части туловища расширена. Подкожный жировой слой развит недостаточно, распределен равномерно. Тургор тканей сохранен. Периферических отеков нет.
В биохимическом анализе крови выявлена ги-пербилирубинемия до 86,8 мкмоль/л (прямой —
74.8 мкмоль/л, непрямой — 12,0 мкмоль/л), повышено содержание трансаминаз (АСТ —
75.9 Ед/л, АЛТ — 81,1 Ед/л), дислипидемия (триглицериды — 2,2 ммоль/л), ГГТ (гамма-глу-тамилтрансфераза) — 201 Ед/л (норма 7-15 Ед/л).
Рис. 2. Общий вид девочки (описание в тексте)
Рис. 3. Очаги гипер- и депигментации на пальцах
УЗИ органов брюшной полости: гепатомега-лия, диффузные изменения паренхимы печени и поджелудочной железы, дискинезия желчного пузыря по гипомоторному типу, холепсамус (скопление желчного песка).
УЗИ органов малого таза: в полости малого таза визуализируется тяж средней эхоплотно-сти размерами 1,1 X 0,3 см, яичники как отдельные структуры не визуализируются; заключение: выраженная гипоплазия внутренних половых органов.
Рентгенография кистей рук: костный возраст соответствует 2,5-3 годам, то есть отстает от календарного на 4-4,5 года.
При ФЭГДС выявлен Нр-ассоциированный поверхностный гастрит, гипертрофический бульбит.
Гормональный статус: ФСГ — 1,59 мМЕ/мл (норма 0,5-3,7 мМЕ/мл), ЛГ — 2,26 мМЕ/мл (норма 0,1-3,8 мМЕ/мл), эстрадиол — 22,9 пг/мл (норма 0-20 пг/мл).
Заключение. Анализ клинико-анамнестиче-ских данных и результатов лабораторно-инстру-ментального обследования в динамике полностью подтверждает трактовку диагноза. Полагаем, что представленное клиническое наблюдение имеет значительный практический интерес, ибо нами впервые описано наличие двух de novo возникших генетических ситуаций у одной больной — моногенного синдрома Алажилля и хромосомной аномалии половых хромосом в виде мозаичного
варианта синдрома Шерешевского—Тернера. Наше наблюдение еще раз доказывает обоснованность своевременного проведения биопсии печени в случае длительного течения синдрома внутрипеченочного холестаза на фоне отсутствия маркеров вирусных гепатитов и эффекта от проводимой терапии с целью верификации диагноза наследственного поражения печени. Кроме того, выраженная задержка роста у девочек, особенно в сочетании с шейным птеригиумом, является несомненным показанием к тщательному ци-тогенетическому обследованию для исключения мозаичного варианта синдрома Шерешевско-го—Тернера. Несомненный клинический интерес представит дальнейшее наблюдение за естественным течением синдрома Алажилля у наблюдаемой нами больной.
1. Alagille D., Odievre M., Gautier M., Dommergues J. P. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental and sexual development, and cardiac murmur // Journal of Pediatrics, St. Louis. — 1975. — Vol. 86. - P. 63-71.
2. Алажилль Д., Одьевр М. Заболевания печени и желчных путей у детей. — М.: Медицина, 1982. - C. 260-265.
3. Джонс К. Л. Наследственные синдромы по Дэвиду Смиту: Атлас-справочник. Пер. с англ. — М., «Практика», 2011. - С. 696-697.
4. Детская гепатология / Под ред. Б. С. Каганова. — М.: Издательство «Династия», 2009. — С. 479-483.
i Не можете найти то, что вам нужно? Попробуйте сервис подбора литературы.5. Мухина Ю. Г., Дегтярева А. В. Холестаз у новорожденных и детей первых месяцев жизни / Детская гастроэнтерология (избранные главы) / Под ред. А. А. Баранова, Е. В. Климанской, Г. В. Римарчук. — М., 2002. — С. 306-352.
7. Шерлок Ш., Дули Дж. Заболевания печени и желчных путей. — М.: ГЭОТАР Медицина, 1999. — 529-531.
8. Oda T., Elkahloun A. G, Pike B. L. et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome // Nat. Genet. — 1997. — Vol. 16. — P. 235-242.
9. Li P.-H., Shu S.-G., Yang C.-H., Lo F.-C., Wen M.-C., Chi C.-S. Alagille syndrome with interstitial 20p deletion derived from maternal ins (7;20) // Am. J. Med. Genet. 1996. — Vol. 63. — P. 537-541.
10. Ghishan F. K., LaBrecque D. R., Mitros F. A. et al. The evolving nature of «infantile obstructive cholangiopathy» // J. Pediat. — 1980. — Vol. 97, № 1. — P. 27-32.
11. Krantz I. D., Piccoli D. A., Spinner N. В. Alagille syndrome. // J. Med. Genet. — 1997. — Vol. 34. — P. 152-157.
12. Bucuvalas J. C., Horn J. A., Carlsson L., Balistreri W. F., Chernausek S. D. Growth hormone insensitivity associated with elevated circulating growth hormone-binding protein in children with Alagille syndrome and short stature // J. Clin. Endocr. Metab. — 1993. — Vol. 76. — P. 1477-1482.
Климов Леонид Яковлевич — к. м. н., доцент кафедры пропедевтики детских болезней и поликлинической педиатрии ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Стоян Марина Валерьевна — студентка VI курса педиатрического факультета ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Вдовина Татьяна Михайловна — к. м. н., врач-генетик АНМО «Ставропольский краевой клинический консультативно-диагностический центр»
Курьянинова Виктория Александровна — ассистент кафедры пропедевтики детских болезней и поликлинической педиатрии ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Погорелова Лариса Витальевна — к. м. н., доцент кафедры детских инфекционных болезней ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Атанесян Роза Артуровна — клинический ординатор кафедры эндокринологии Института последипломного образования ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Долбня Светлана Викторовна — студентка V курса педиатрического факультета ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Кулешова Ольга Констатнтиновна — к. м. н., доцент кафедры пропедевтики детских болезней и поликлинической педиатрии ГБОУ ВПО «Ставропольская государственная медицинская академия» Минздравсоцразвития РФ
Еремеева Ольга Ильинична — заведующий гастроэнтерологическим отделением Ставропольской детской городской клинической больницы имени Г. К. Филиппского, врач высшей категории
Углова Татьяна Алексеевна — заведующий эндокринологическим отделением Ставропольской детской городской клинической больницы имени Г. К. Филиппского, врач высшей категории
Кадурина Тамара Ивановна — д. м. н., профессор кафедры медицинской генетики ГОУ ДПО «Санкт-Петербургская медицинская академия последипломного образования» Минздравсоцразвития РФ
Синдром Алажиля ( Артериопеченочная дисплазия )
Синдром Алажиля — это редкое генетическое заболевание, при котором нарушения гепатобилиарной и сердечно-сосудистой системы сочетаются с аномалиями развития скелета. Характерные симптомы проявляются у ребенка с первых месяцев жизни и включают желтуху, кожный зуд, нутритивную недостаточность, отставание темпов психомоторного развития. Для диагностики синдрома проводят УЗИ и биопсию печени, эхокардиографию, биохимическое и генетическое тестирование. Лечение болезни Алажиля состоит из диетотерапии, приема желчегонных и антигистаминных медикаментов. По показаниям назначается хирургическое вмешательство.
Осложнения
Обострения холестаза при синдроме Алажиля зачастую сопровождаются инфекцией, что провоцирует холецистохолангит, осложняющий течение болезни. Повреждение печени желчными кислотами и хроническое воспаление приводят к перипортальному фиброзу, а у 15% детей уже в раннем возрасте формируется цирроз печени, что является прогностически неблагоприятным признаком.
На фоне хронической мальабсорбции и мальдигестии возникают белково-энергетическая недостаточность и авитаминозы. Тяжелая задержка роста и физического развития отмечаются у 50-87% детей. Также появляются сердечно-сосудистые осложнения: без своевременной коррекции кардиальный порок быстро прогрессирует. 6-летняя выживаемость пациентов с внутрисердечными поражениями составляет до 40%, а без таковых — повышается до 95%.
Аннотация научной статьи по клинической медицине, автор научной работы — Климов Л.Я., Стоян М.В., Вдовина Т.М., Курьянинова В.А., Погорелова Л.В.
В статье впервые в литературе представлено наблюдение аутосомно-доминантно наследуемого синдрома Алажилля в сочетании с мозаичным вариантом синдрома Шерешевского Тёрнера у девочки 7 лет. Диагностировано столь редкое сочетание благодаря проведению биопсии печени у ребёнка с синдромом внутрипечёночного холестаза и цитогенетического обследования в связи с отставанием в росте и множественными дизморфиями.
Патогенез
Ведущим патогенетическим компонентом является сужение внутрипеченочных желчных протоков, затрудняющее отток желчи с ее последующим накоплением в гепатоцитах. Желчные кислоты оказывают токсическое влияние на печеночные клетки, вызывая нарушение их функций и гибель, а также проникают в системный кровоток, провоцируя кожные признаки синдрома. Вследствие недостаточного поступления желчи в кишечник возникает мальабсорбция.
В механизме развития симптоматики важную роль играют сердечные пороки, которые сопровождаются нарушениями кровообращения и гипоксией тканей. Нарушения гемодинамики связаны с отсутствием выброса крови из правого желудочка и гипотрофией миокарда. При несвоевременной диагностики происходит дилатация правого предсердия и желудочка. При тяжелой форме синдрома может быть вторичное открытие овального окна и венозно-артериальный сброс крови.
Синдром Алажилля у девочки с синдромом Шерешевского-Тернера Текст научной статьи по специальности «Клиническая медицина»
Причины
Синдром Алажиля — наследственное заболевание, которое передается по аутосомно-доминантному типу. Он вызван мутацией на коротком плече 20 хромосомы, где расположен ген Jagged 1 (JAG1). Учитывая тип наследования, патология с одинаковой частотой встречается у мальчиков и девочек, может повторяться в каждом поколении семьи. Описаны случаи развития синдрома у младенцев с трисомией по 21 хромосоме (болезнью Дауна).
Синдром Алажилля – причины, диагностика, проявления и рекомендации
В 1975 году французский педиатр-гепатолог впервые описал группу детей с холестатическим заболеванием печени, которое демонстрировало и другие особенности, включая проблемы с сердцем и характерные черты лица. Это расстройство было названо синдромом Алажилля-Ватсона, синдромом недостаточности желчного протока и артериопеченочной дисплазией, но наиболее оно известно как синдром Алажилля.
Значительный вклад в понимание синдрома Алажилля внесли врачи и ученые детской больницы Филадельфии. В 1997 году был идентифицирован ген, вызывающий синдром Алажилля, который коренным образом изменил возможность диагностировать заболевание и консультировать семьи. С момента первоначального открытия было изучено более 200 пациентов, чтобы узнать, какие типы генных изменений приводят к заболеванию и как работают эти гены. Недавно был идентифицирован второй ген, который также способен вызывать синдром Алажилля.
Если вашему ребенку был поставлен диагноз синдрома Алажилля, у вас, вероятно, есть бесчисленные вопросы о том, что это такое, что его вызывает, чего ожидать и какими будут симптомы у вашего ребенка. На многие из этих вопросов мы ответим здесь, но, пожалуйста, поговорите с врачом вашего ребенка или другим специалистом в области здравоохранения.
Что такое синдром Алажилля?
Синдром Алажилля, также известный как синдром Алажилля-Ватсона, синдром малокровия желчных протоков и артериопеченочная дисплазия, является аутосомно-доминантным наследственным заболеванием, с которым связаны нарушения со стороны печени, сердца, глаз и скелета, а также характерные черты лица.
Характерные черты лица:
- выдающийся лоб и остроконечный подбородок, придающий лицу вид треугольника;
- глубоко посаженные глаза;
- прямой нос.
Проблемы с печенью:
- холестаз (затрудненный отток желчи из печени);
- желтуха, вызванная неспособностью печени правильно обработать желчь;
- слишком малое количество желчных протоков (внутрипеченочная недостаточность желчных протоков), которое обнаруживается при биопсии печени.
Врожденные кардиологические аномалии:
- периферический легочный стеноз (сужение легочной артерии);
- сердечные шумы;
- любой порок сердца, хотя чаще обнаруживаются дефекты правой стороны сердца, например, тетрада Фалло.
Аномалии органа зрения:
- задний эмбриотоксон;
- аномалия Аксенфельда.
Почечные аномалии:
- почечный тубулярный ацидоз;
- структурные аномалии почек.
Скелетные аномалии:
- бабочковидные позвонки;
- уменьшение межпозвонкового расстояния;
- низкий рост.
Причины
Синдром Алажилля в большинстве случаев вызван изменениями в гене, названном Jagged1, расположенным на хромосоме 20. В 3-5% случаев весь ген удаляется (отсутствует) из одной копии хромосомы 20. Причиной заболевания являются изменения или мутации в последовательности ДНК, которая составляет ген Jagged1. В очень небольшом числе случаев, менее 1%, к этому синдрому приводят изменения в другом гене, Notch 2.
Около трети детей с синдромом Алажилля наследуют изменение в Jagged1 от родителя. В остальных двух третях мутация в Jagged1 впервые появляется у этого ребенка. Синдром Алажилля является аутосомно-доминантной патологией, что означает, что у человека, несущего мутацию гена Jagged1, есть 50%-ый шанс передать эту мутацию своему ребенку.
Эффект от наличия мутации в Jagged1 может сильно различаться. Некоторые люди, наследующие мутацию, имеют тяжелый синдром Алажилля, включающий болезни сердца и печени, в то время как у других проявления незначительны, например, задний эмбриотоксон или характерные черты лица.
Диагностика
Когда первоначально был описан синдром Алажилля, для подтверждения диагноза требовалось, чтобы у человека была недостаточность желчного протока в дополнение к по меньшей мере трем из пяти основных критериев:
- холестаз;
- характерные черты лица;
- вертебральные аномалии;
- нарушения со стороны органа зрения;
- пороки сердца.
Из-за успешной идентификации Jagged1 как гена, вызывающего синдром Алажилля, диагностические критерии были изменены. В последние годы было принято, что диагноз может быть поставлен человеку, у которого нет всех клинических критериев синдрома Алажилля, но есть мутация в Jagged1. Например, считается, что менее тяжело затронутый член семьи, который не соответствовал бы клиническим критериям, но имеет ту же мутацию Jagged1, что и сильно пострадавший член семьи, имеет синдром Алажилля.
Поскольку проявления и тяжесть синдрома Алажилля могут широко варьироваться, его часто бывает трудно диагностировать.
Функция печени
Большинство людей с синдромом Алажилля имеют заболевание печени, это может варьироваться в зависимости от его тяжести, а также изменяться со временем. У детей в раннем периоде обычно развивается желтуха, и это наиболее распространенное время для оценки гастроэнтерологом.
Первоначальная диагностика заболевания печени будет включать в себя физическое обследование, при котором особое внимание уделяется увеличению печени и/или селезенки. Функция печени оценивается с помощью анализов крови, которые называются печеночными пробами.
В случае необходимости могут потребоваться дальнейшие исследования. Это может быть УЗИ брюшной полости с целью исключения других причин желтухи и оценки степени увеличения печени и селезенки. Иногда помогает диагностировать синдром Алажилля биопсия печени, хотя она не обязательна для постановки диагноза.
Тесты на функцию поджелудочной железы
Врачи могут косвенно оценить функцию поджелудочной железы с помощью 72-часового сбора стула в сочетании с трехдневной записью диеты, чтобы определить, сколько жира ребенок поглощает из пищи. Сбор обычно выполняется дома, предоставляются необходимые материалы и инструкции. Все испражнения в течение 72 часов собирают в предварительно взвешенные контейнеры и замораживают. Все, что ребенок ест в течение этих трех дней, нужно взвешивать и записывать. Затем лаборатория определяет количество потребляемого жира (коэффициент поглощения жира). К сожалению, этот тест не может отличить мальабсорбцию жира из-за заболевания печени от мальабсорбции жира из-за заболеваний поджелудочной железы.
Кардиология
Ваш ребенок будет обследован кардиологом, чтобы выявить любые проблемы с сердцем.
Кардиолог выполнит полное физическое обследование и, скорее всего, ЭКГ и эхокардиограмму. ЭКГ измеряет сердечный ритм. Эхокардиограмма является ультразвуковым исследованием сердца, она определяет точную структуру и функцию сердца. В зависимости от результатов кардиолог может принять решение назначить дополнительные тесты или организовать визиты в будущем.
Офтальмология
У многих детей с синдромом Алажилля есть аномалия органа зрения. Наиболее распространен задний эмбриотоксон, дополнительная круговая линия на поверхности глаза, которая не приводит к каким-либо нарушениям зрения. Требуется специализированный осмотр для обнаружения этой аномалии. У некоторых детей с синдромом Алажилля присутствуют пигментные изменения сетчатки (тонкий слой фоторецепторов, выстилающих внутреннюю стенку глаза). Офтальмолог исследует глаза вашего ребенка с целью выявления заднего эмбриотоксона и других аномалий.
Рентгенодиагностика
Рентген позвоночника помогает обнаружить бабочковидные позвонки, наиболее распространенный скелетный дефект, связанный с синдромом Алажилля. Аномалия, из-за которой позвонки расщепляются, дает появление «летающей бабочки» на рентгеновских снимках. Как правило, бабочковидные позвонки не вызывают никаких симптомов, но помогают в постановке диагноза.
Генетическая диагностика
Если врач вашего ребенка подозревает у него синдром Алажилля, ребенок должен быть проконсультирован генетиком и генетическим консультантом. В частности, генетик сможет оценить черты лица, характерные для синдрома.
Вместе генетик и генетический консультант рассмотрят результаты других диагностических исследований, а также вашу семейную историю. Они предоставят информацию о генетике и схеме наследования синдрома Алажилля. Основываясь на информации, полученной из истории вашей семьи и результатов генетического тестирования, они также смогут ответить на вопрос о вероятности того, что будущие дети в вашей семье могут пострадать от синдрома Алажилля.
Проблемы роста и питания при синдроме Алажилля
У детей с синдромом Алажилля распространены низкорослость, задержка полового созревания и недоедание. Дети с синдромом Алажилля имеют больший недостаток роста, чем дети с другими хроническими заболеваниями печени, такими как атрезия желчных путей. Хотя точная причина этого неизвестна, возможны следующие:
- жировая мальабсорбция (неспособность поглощать жир);
- недостаточное потребление калорий или питательных веществ;
- дефект гена Jagged1.
Правильное питание для детей с синдромом Алажилля необходимо для предотвращения и лечения низкорослости, а также для предотвращения и лечения дефицита питания. Дети с синдромом могут оцениваться и контролироваться врачом-диетологом для оценки их роста и состояния питания, а также для получения индивидуальных рекомендаций по диете. Детям, которые не могут потреблять достаточно калорий может потребоваться назогастральная трубка или гастростомическая трубка для улучшения их питания и роста.
У детей с синдромом Алажилля, которые перенесли трансплантацию печени, происходит не так много «догоняющего» роста после трансплантации, как у детей с другими заболеваниями печени, что может свидетельствовать о том, что низкий рост обусловлен первичным дефектом гена в Jagged1. В отличие от других педиатрических заболеваний печени, при синдроме Алажилля нарушения роста могут затронуть системы органов, например, почки или сердце.
Врач вашего ребенка и диетолог будут работать с ребенком, чтобы гарантировать ему наилучшее питание для обеспечения роста и развития. Важно, чтобы дети с синдромом Алажилля потребляли достаточно калорий и поддерживали нормальный уровень витаминов в сыворотке.
Витаминные добавки для детей с синдромом Алажилля
Большинство детей с синдромом Алажилля должны принимать витаминные добавки. Поскольку у таких детей возникают трудности с поглощением жира, у них может развиться дефицит жирорастворимых витаминов (A, D, E и K), если они не получат надлежащих добавок. Им также может потребоваться дополнительный кальций и цинк. Лучший способ для ребенка с синдромом Алажилля получать витамины, которые ему нужны, это прием отдельных для каждого витамина витаминных добавок. Следует контролировать уровень содержания витаминов в крови и корректировать их прием соответствующим образом. Обязательно поговорите с врачом вашего ребенка о том, какие витаминные добавки могут понадобиться. Кроме назначенных, не давайте ребенку никаких дополнительных витаминов без предварительной консультации с врачами, потому что, например, слишком большое количество витамина А может быть токсичным для печени.
Диета для детей с синдромом Алажилля
Дети с синдромом Алажилля должны потреблять высококалорийное питание из-за плохого роста и снижения способности абсорбировать жир. Ребенок может выделять до 50% употребленного жира с калом. Врач в этом случае может назначить пищевую добавку, содержащую триглицериды средней цепи, потому что дети с синдромом Алажилля лучше поглощают этот тип жира.
Некоторые родители говорят, что из-за продуктов с высоким содержанием жиров у ребенка появляется рыхлый маслянистый стул, поэтому они сажают своего ребенка на диету с низким содержанием жиров. Этого делать не рекомендуется, поскольку жир является наиболее концентрированным источником калорий. Если ребенок вынужден находиться на диете с низким содержанием жира, потому что он не переносит жир, необходимо приложить усилия, чтобы ребенок потреблял достаточно калорий, чтобы обеспечить рост. Диетолог поможет оценить рост и диету ребенка, а затем дать рекомендации по питанию.
Проблемы развития и синдром Алажилля
Общие сведения
Синдром Алажиля (артериопеченочная дисплазия) занимает второе место (17%) среди всех заболеваний гепатобилиарного тракта у детей в первые 6 месяцев после рождения. Он выявляется с частотой 1 случай на 70 тыс. новорожденных. Патология получила свое название по имени французского педиатра Даниеля Алажиля, который подробно описал ее в 1975 году. В медицинской литературе болезнь также известна как артериопеченочная дисплазия. Полиморфизм симптоматики, сложность диагностики и серьезный прогноз обуславливают значимость синдрома в неонатологии и детской гастроэнтерологии.
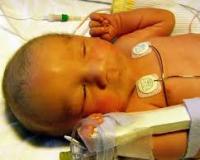
Диагностика
Опытный неонатолог или педиатр может заподозрить синдром Алажиля по клиническим проявлениям, но при умеренной выраженности симптомов и их позднем развитии постановка диагноза сопровождается трудностями. При физикальном обследовании врач выявляет гепатомегалию, сердечный горб, грубый систолический шум во 2 межреберье. Для подтверждения синдрома проводятся инструментальные и лабораторные исследования:
- УЗИ органов брюшной полости. Скрининговое неинвазивное исследование выполняется для визуализации печени и определения степени гепатомегалии. Сонография необходима, чтобы исключить другие причины желтухи, оценить состояние гепатобилиарной системы и желудочно-кишечного тракта ребенка.
- Эхокардиография. При визуализации сердца врач определяет сужение легочной артерии на уровне клапанов или подклапанного пространства, гипертрофию правого желудочка, дилатацию правых камер сердца. При необходимости производится рентгенологическая диагностика, которая показывает выступающую дугу легочной артерии, расширение границ сердца.
- Биопсия печени. Снижение числа внутрипеченочных протоков — патогномоничный признак синдрома Алажиля, но он может не обнаруживаться в младенческом возрасте. Биопсия рекомендована для исключения других гепатобилиарных патологий, если детский гастроэнтеролог испытывает трудности с дифференциальной диагностикой.
- Анализ крови. При биохимическом исследовании определяют повышение типичных маркеров холестаза — холестерина, щелочной фосфатазы, прямого билирубина. У ребенка наблюдается дислипопротеинемия. Ферменты цитолиза в печеночных пробах повышаются умеренно. Отклонения в протеинограмме у большинства пациентов отсутствуют.
- Генетическое тестирование. Молекулярная диагностика мутаций гена JAG1 требуется при мягких признаках синдрома или его атипичном протекании. При прямом секвенировании характерные генные нарушения удается обнаружить у 95% детей, но оно делается только на научно-исследовательской основе.
Прогноз и профилактика
Успех лечения зависит от степени выраженности синдрома: при незначительной артериопеченочной дисплазии удается компенсировать состояние и обеспечить нормальное развитие ребенка, а тяжелые формы болезни Алажиля коррелируют с необратимыми повреждениями печени, кардиоваскулярными расстройствами. Профилактика патологии заключается в медико-генетическом консультировании пары на этапе планировании беременности.
Лечение синдрома Алажиля
Консервативная терапия
Для коррекции нутритивной недостаточности ребенку назначают усиленное лечебное питание с ограничением длинноцепочечных жиров и повышением процента среднецепочечных, которые лучше усваиваются при дефиците желчи. В ежедневном рационе повышают содержание жирорастворимых витаминов. При выраженных расстройствах питания специальные составы вводятся через назогастральный зонд. Медикаментозное лечение включает:
- Желчегонныепрепараты. Необходимы для стимуляции оттока желчи из печеночных протоков, что способствует облегчению желтухи, улучшению пищеварения и устранения дискомфорта. Хороший эффект на гепатобилиарную систему оказывает урсодезоксихолевая кислота (УДХК).
- Антигистаминные средства. Показаны для облегчения мучительного кожного зуда у детей, однако они не всегда эффективны. При рефрактерных случаях терапию дополняют седативными средствами, транквилизаторами.
- Витамины и минералы. Для нормализации системы свертываемости крови эффективен витамин К в пероральной или парентеральной форме. Для улучшения кальциево-фосфорного обмена применяются препараты холекальциферола, а при снижении плотности костной ткани используют добавки с кальцием.
Хирургическое лечение
При кардиальных аномалиях выполняются малоинвазивные методы коррекции — вальвулопластика, баллонное расширение просвета легочной артерии и ее стентирование. При тяжелых комбинированных пороках рекомендованы открытые кардиохирургические операции. При циррозе, печеночной недостаточности, хронической портальной гипертензии рассматривается вопрос о трансплантации печени.
Симптомы
Для классического синдрома Алажиля характерно сочетание 5 признаков: холестаз, стеноз легочной артерии или другие кардиальные пороки, нарушения формирования лицевого скелета, поражения глаз, незаращение тел позвонков. Для постановки диагноза достаточно наличия 3-х симптомов из этого списка. Во всех случаях развивается поражение печени, а на втором месте по частоте стоят сердечно-сосудистые патологии (до 87-95%).
Клинические признаки синдрома проявляются в первые 3 месяца жизни младенца, при менее грубых аномалиях начало болезни приходится на второе полугодие жизни. Первым симптомом является умеренная желтушность кожи, иногда с зеленоватым оттенком. Она дополняется мучительным кожным зудом, из-за чего младенец становится беспокойным, постоянно плачет, плохо спит.
Расстройства продукции и выделения желчи сопровождаются диспепсическими нарушениями, обесцвечиванием кала, но этот признак появляется намного позже желтухи. Нарушения липидного обмена могут спровоцировать появление ксантом — доброкачественных образований желтоватого цвета, которые возвышаются над уровнем кожи. В основном они локализованы на пальцах, по задней поверхности шеи, в паховой и подколенной зонах.
Для больных, страдающих синдромом Алажиля, специфичны особенности внешности, обусловленные лицевым дизморфизмом. Родители и врачи обращают внимание на слишком широкий лоб, глубоко посаженные и широко расставленные глаза, длинный прямой нос. У ребенка могут быть оттопыренные и низко расположенные уши, выступающий подбородок. Вследствие офтальмопатии у младенца возникают затруднения с узнаванием родителей, он не интересуется яркими игрушками.
МКБ-10
Alagille Syndrome

Alagille Syndrome is an inherited disorder that closely resembles other forms of liver disease seen in infants and young children. However, a group of unusual features affecting other organs distinguishes Alagille Syndrome from the other liver and biliary diseases of infants.
Facts at a Glance
- Alagille Syndrome occurs in about one of every 30,000 live births. The disorder affects both sexes equally and shows no geographical, racial, or ethnic preferences.
- A person with Alagille Syndrome has fewer than the normal number of small bile ducts inside the liver.
Information for the Newly Diagnosed
What are the symptoms of Alagille Syndrome?
Symptoms of Alagille Syndrome are jaundice; pale, loose stools; and poor growth within the first three months of life. Later, there is persistent jaundice, itching, fatty deposits in the skin, and stunted growth and development during early childhood. The disease often stabilizes between ages four and ten with an improvement in symptoms.
Other features, which help establish the diagnosis, include abnormalities in the kidneys, cardiovascular system, eyes, and spine. Narrowing of the blood vessel connecting the heart to the lungs leads to heart murmurs but rarely causes problems in heart function. The shape of the bones of the spinal column may look like the wings of a butterfly on x-ray, but this shape almost never causes any problems with function of the nerves in the spinal cord.
More than 90% of children with Alagille syndrome have an unusual abnormality of the eyes. An extra, circular line on the surface of the eye can be detected during a specialized eye examination. In addition, some children may have some changes in kidney function.
Many physicians believe that there is a specific facial appearance shared by most of the children with Alagille syndrome that makes them easily recognizable. The features include a prominent, broad forehead; deep-set eyes; a straight nose; and a small pointed chin.
What causes Alagille Syndrome?
Alagille Syndrome is a genetic condition (associated with the Notch signaling pathway and Jagged1 gene) that causes narrowed and malformed bile ducts in the liver. Bile that cannot flow through the deformed ducts builds up in the liver and causes scarring. The scar tissue prevents the liver from working properly to eliminate wastes from the bloodstream.
How is Alagille Syndrome diagnosed?
A diagnosis of Alagille Syndrome usually depends upon finding several different components of the syndrome in an individual. Generally, the syndrome involves five distinct findings, including reduced bile flow, congenital heart disease, bone defects, a thickening of a line on the surface of the eye, and particular facial features. Diagnosis can be confirmed by genetic analysis.
How is Alagille Syndrome treated?
Treatment of Alagille Syndrome focuses on trying to increase the flow of bile from the liver, maintaining the child’s normal growth and development pattern, and correcting any of the nutritional deficiencies that often develop. Because bile flow from the liver to the intestine is slowed in Alagille syndrome patients, medications designed to increase the flow of bile are frequently prescribed.
While reduced bile flow into the intestine leads to poor digestion of dietary fat, a specific type of fat can still be well digested, and therefore infant formulas containing high levels of medium-chain triglycerides (MCT) are usually substituted for conventional formulas. Some infants can grow adequately on breast milk if additional MCT oil is given. There are no other dietary restrictions.
Sometimes surgery is necessary during infancy to help establish the diagnosis of Alagille syndrome by direct examination of the bile duct system and through liver biopsy. However, surgical reconstruction of the bile duct system is not recommended because bile can still flow from the liver, and there is presently no procedure that can correct for the loss of the bile ducts within the liver. Occasionally, liver cirrhosis advances to a stage where the liver fails to perform its functions. Liver transplantation is then considered.
Who is at risk for Alagille Syndrome?
Alagille syndrome is generally inherited only from one parent and there is a 50% chance that each child will develop the syndrome. The genetic basis has recently been defined and the “Alagille gene” has been found. Each affected adult or child may have all or only a few of the features of the syndrome. Frequently, a parent, brother, or sister of the affected child will share the facial appearance, heart murmur, or butterfly vertebrae, but have a completely normal liver and bile ducts.
Похожие темы научных работ по клинической медицине , автор научной работы — Климов Л.Я., Стоян М.В., Вдовина Т.М., Курьянинова В.А., Погорелова Л.В.
Врожденная гипоплазия желчных протоков (синдром аллажила-уотсона) — редкая причина билиарного цирроза у взрослых Первый клинический случай Синдрома Алажиля в Азербайджане Эхографические изменения органов брюшной полости у детей с билиарной атрезией и синдромом Алажиля в течение первых 3 месяцев жизни i Не можете найти то, что вам нужно? Попробуйте сервис подбора литературы.Читайте также: