
Fragile x syndrome что это
Обновлено: 19.04.2024
Полвека назад возникла и начала стремительно развиваться молекулярная биология. Биологи и физики (обычный для середины двадцатого века союз) открыли важнейшие клеточные процессы, изобрели основные методы, без которых сегодня немыслима работа любой биологической лаборатории. Сейчас у нас есть громадный потенциал для решения всевозможных задач: прояснения аспектов происхождения жизни, изучения взаимодействий компонентов в живой клетке и сложных биохимических каскадов. Мы знаем и умеем то, что еще лет 60 назад казалось фантастикой. И одна из задач, на решение которой уже могут покуситься ученые, – это борьба с наследственными заболеваниями человека. Некоторые из них, такие как фенилкетонурия, успешно корректируются, подходы к терапии множества других еще не найдены. В этой статье пойдет речь об одном из таких заболеваний – синдроме ломкой X-хромосомы – и о сложностях его изучения.
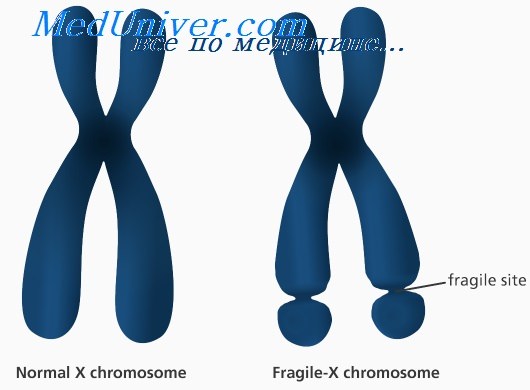
В начале 20 века ученые заметили, что умственная отсталость чаще поражает мужчин. В 1934 году ирландский врач Джеймс Мартин и английский генетик Джулия Белл впервые описали семью, где умственная отсталость наследовалась сцепленно с полом. В этой семье было 11 мужчин-олигофренов и две женщины с легкой степенью умственной отсталости. Обнаруженную семейную форму заболевания назвали синдромом Мартина-Белл. Через 35 лет Герберт Лабс, проводя цитогенетическое исследование, выявил в кариотипе четырех слабоумных мужчин и трех нормальных женщин (из трех поколений одной семьи) странную Х-хромосому, которую он назвал маркерной: ближе к концу длинного плеча у нее была вторичная перетяжка. Лабс предложил отслеживать маркерную хромосому у эмбрионов мужского пола в неблагополучных по синдрому Мартина-Белл семьях, поскольку она может сигнализировать о высоком риске рождения олигофренов (рис. 1) [1]. Так медики обрели первый пренатальный маркер синдрома, а их пациенты – возможность принятия взвешенного решения о сохранении беременности. Маркерную перетяжку локализовали на участке Xq27.3.

Рисунок 1. Герберт Лабс рассказывает коллегам о перетяжке на X-хромосоме при синдроме Мартина-Белл. Здесь и ниже рисунки автора статьи.
Позже многие исследователи наблюдали под микроскопом Х-хромосомы не просто с перетяжкой, а как бы поломанными – с «оторванными» кончиками длинных плеч. Место перетяжки/поломки стали называть ломким сайтом (fragile site). Поэтому заболевание получило другое название – синдром ломкой X-хромосомы (fragile X syndrome).
Еще одна интересная особенность этого заболевания – усугубление болезни в каждом последующем поколении (генетическая антиципация). Этот феномен объяснили только в последнем десятилетии 20 века, после открытия особого типа мутаций – экспансии тринуклеотидных повторов.
Если классифицировать заболевания по патогенетическим механизмам, то довольно большую группу составят болезни, обусловленные экспансией повторов [2]. Суть мутации заключается в следующем: в человеческом геноме встречаются короткие участки (например, триплеты нуклеотидов), в норме повторенные несколько раз, но по каким-то причинам их количество начинает резко расти – в десятки и сотни раз, – и общая длина «заикающегося» (содержащего повторы) фрагмента может увеличиться до нескольких тысяч пар нуклеотидов (рис. 2).

Рисунок 2. Представим, что наш геном – очень длинный текст, а происходящая экспансия превращает его в полную бессмыслицу.
Позже выяснилось, что экспансия лежит в основе патогенеза не только синдрома ломкой X-хромосомы, но и миотонической дистрофии I и II типов, а также ряда нейродегенеративных заболеваний человека – например, бокового амиотрофического склероза и болезни Гентингтона. В общей сложности известно около 30 заболеваний, для которых характерна такая мутация. Многие из этих патологий связаны с увеличением количества повторов (ЦГГ)n, (ЦАГ)n, (ГАА)n и других [3].
Причины и патогенез синдрома ломкой X-хромосомы
Синдром ломкой X-хромосомы, пожалуй, самая распространенная причина наследственной умственной отсталости после синдрома Дауна. Клинических проявлений синдрома довольно много и наблюдаются из них не все и не всегда, но основные – низкий уровень интеллекта и эмоционального развития вкупе с рядом физических отклонений, – присутствуют чаще всего. Эти особенности заметны уже в раннем детстве.
Причина заболевания кроется в увеличении числа повторов триплета ЦГГ в области промотора (стартовой площадки для начала синтеза мРНК) гена FMR1 (рис. 3). Продуктом этого гена является белок FMRP (fragile X mental retardation protein), который взаимодействует с РНК и направляет сложные молекулярные каскады, необходимые для нормального формирования нейронов, их синаптической пластичности [4]. У здорового человека количество повторов варьирует в пределах от 5 до 54. При увеличении числа повторов до 55–200 возникает аллель, называемый премутантным. В популяции он встречается достаточно часто: у одного из 200–250 человек. Хотя уровень мРНК гена оказывается выше нормы, содержание FMRP остается неизменным или даже немного снижается. Почему это происходит – пока неизвестно. Можно предположить, что в этом участвует РНК-интерференция – процесс подавления экспрессии гена (какого-то этапа пути от нуклеотидной последовательности до конечного продукта, в данном случае – FMRP) с помощью малых РНК [5].

Рисунок 3. Структура гена FMR1 и схема его экспрессии. 5’-НТО и 3’-НТО – 5’- и 3’-нетранслируемые области гена.
При чуть более выраженной экспансии ЦГГ-повторов у пациентов можно обнаружить особые внутриклеточные включения, состоящие из мРНК FMR1 и РНК-связывающих белков. Это свидетельство того, что мРНК становится токсичной для клетки [6]. Интересно, что «нормальная» мРНК не оказывает токсического эффекта даже в очень высоких концентрациях. У большинства женщин – носительниц премутации, в отличие от мужчин, нет внешних проявлений патологии. В этом заслуга второй X-хромосомы, которая в большей или меньшей доле клеток компенсирует дефект. Более того, есть данные о преимущественной инактивации («выключении») именно дефектной хромосомы. Но зачастую таким женщинам свойственны эмоциональные проблемы, депрессии и фобии.
Инактивация одной из Х-хромосом – жизненно важный процесс дозовой компенсации генов, препятствующий удваиванию экспрессии всех Х-хромосомных генов у самок по сравнению с самцами. То есть в каждой клетке особи любого пола, несмотря на диплоидный набор хромосом, активна только какая-то одна X-хромосома – доставшаяся либо от отца, либо от матери. О захватывающих подробностях «выключения» половых хромосом у человека и червя рассказывают статьи «Загадочное путешествие некодирующей РНК Xist по X-хромосоме» [8] и «Истории из жизни Х-хромосомы круглого червя-гермафродита» [9]. – Ред
И, разумеется, даже при отсутствии внешних признаков заболевания премутантный аллель передается потомству. При этом происходит «амплификация» повтора – с каждым овогенезом всё больше и больше, вплоть до нескольких тысяч «копий». А это ведет к тому, что премутантный аллель превращается в самый что ни на есть мутантный [2, 7]. В этом случае мы говорим уже о синдроме ломкой Х-хромосомы. Его частота в популяции составляет около 1:3600–6000. Это довольно много! При таком значительном увеличении количества повторов ЦГГ происходят эпигенетические изменения: присоединение метильных групп к цитозину ЦГГ-триплетов в области промотора FMR1 и модификации белков, связанных с ДНК, – гистонов. Всё это ведет к локальному изменению плотности укладки ДНК – формированию конденсированного, неактивного хроматина, называемого гетерохроматин. Экспрессия генов, находящихся в такой зоне, подавляется. Поэтому в случае синдрома Мартина-Белл резко сокращается продукция белка FMRP. Более того, модификации хроматина вызывают визуальную «ломкость» хромосомы в районе Xq27 – ту самую, которую наблюдали ученые еще в середине 20 века. Правда, справедливости ради надо отметить, что у пары процентов больных синдром обусловлен не экспансией ЦГГ-повторов, а другими мутациями гена FMR1.
Итак, судя по всему, патогенетические механизмы у синдрома ломкой X-хромосомы и других «экспансивных» заболеваний общие: для всех них характерно какое-то критическое количество триплетов, при котором ген еще функционирует нормально. Причины самόй экспансии до конца не ясны. На сегодняшний день предложено множество гипотез и моделей, пытающихся ее объяснить, например, нарушениями при репликации, проблемами с системами репарации, и т.д. Однако пока ни одна из них не нашла экспериментального подтверждения.
Почему сложно диагностировать экспансию и как эту проблему решают?
Как уже упоминалось, синдром ломкой X-хромосомы далеко не единственное заболевание, проявляющееся умственной отсталостью. Но накопленные знания помогли разработать достаточно подробную методику диагностики именно этого синдрома. Есть возможность выявить даже премутацию у людей с нормальным фенотипом (с нормальным уровнем IQ и без аномалий развития) [10]. Это очень важно, поскольку у женщин-носительниц высок риск появления детей с выраженным синдромом. Правда, эта методика не лишена недостатков и, к сожалению, не применяется широко, поэтому разработке методов молекулярной диагностики до сих пор отводится особое место.
Первоначально проводили исследование хромосомного набора пациента – кариотипирование, – и при обнаружении повреждений в участке Xq27.3 ставили диагноз. Это и сегодня является первым, что делают врачи-генетики – по крайней мере, в России. Проблема кариотипирования заключается в том, что этот метод недостаточно чувствителен, а значит, не слишком надежен. Поэтому всё чаще для постановки диагноза применяют более современные методы. Существуют тест-системы для ДНК-диагностики, основанные на ключевых методах молекулярной биологии: ПЦР (рис. 4), Саузерн-блоте, иммунопреципитации и др. Они позволяют оценить количество белка FMRP и его мРНК, определить число ЦГГ-повторов и уровень метилирования цитозина в промоторе гена FMR1. Это, в свою очередь, помогает лучше понять патофизиологию синдрома, потому что можно соотнести результаты анализа с фенотипом пациентов и носителей премутации.

Рисунок 4. ПЦР – полимеразная цепная реакция, один из стандартных методов молекулярной биологии, применяемых в диагностике. Показаны основные компоненты, без которых реакция не пойдет. ДНК-матрица – молекула ДНК, участок которой нужно многократно размножить (амплифицировать). Праймеры – олигонуклеотиды, комплементарные концам (на разных цепях) интересующего участка ДНК-матрицы, как бы ограничивающие его, – выполняют функцию затравки для фермента, копирующего ДНК (ДНК-полимеразы). дНТФ – дезоксирибонуклеозидтрифосфаты – строительный материал для новой молекулы ДНК. Буфер – раствор солей, обеспечивающий необходимые условия (pH, ионную силу); он обязательно содержит соль магния, потому что только в присутствии ионов Mg2 + работает ДНК-полимераза. Если все компоненты смешать, поместить в прибор под названием амплификатор (циклер) и запустить нужную программу циклического повышения-снижения температуры, на матрице исходных единичных молекул ДНК синтезируются тысячи копий интересующего участка, который в итоге будет легко изучать. Если же из-за каких-то мутаций изменятся последовательности, в норме комплементарные праймерам, либо радикально увеличится расстояние между ними, ПЦР-продукта просто не будет.
ПЦР – основной метод диагностики. Он позволяет наработать область, содержащую (ЦГГ)n. Проведя такой анализ, можно установить точный размер этой области, а значит, и число повторов, и таким способом обнаружить у пациентов премутантные или мутантные аллели. Но нужно сказать, что достичь этого непросто. Исследователи сталкиваются с рядом сложностей при амплификации этих фрагментов. У ДНК, которая будет выступать матрицей для синтеза новых молекул, есть такая характеристика, как ГЦ-состав, отражающая, насколько матрица богата пáрами гуанин-цитозин (богатая матрица содержит примерно 60% ГЦ-пар). Если процент ГЦ-пар высокий, то молекула будет тугоплавкой, и на некоторых этапах ПЦР нужно будет проводить более длительную денатурацию. (ЦГГ)n-область на 100% состоит из пар ГЦ, и ясно, что это очень трудная матрица.
Всё еще более усложняется тем, что такая последовательность без особых усилий образует различные вторичные структуры, очень устойчивые термодинамически: всевозможные шпильки, G-квадруплексы (четыре цепи, связанные между собой гуанинами и поддерживающиеся одновалентным катионом, например K+), i-мотивы (структуры, состоящие из четырех цепей ДНК, богатых цитозином, стабильные в кислой среде) [11]. Изучение подобных структур – очень красивая и интригующая задача для биохимиков и биофизиков, но для установления размера (ЦГГ)n-областей – это серьезное препятствие. Ну и ко всему прочему праймеры (олигонуклеотидные затравки для ДНК-полимеразы) могут образовывать с такими последовательностями димеры, и смесь молекул превращается в один термостабильный нераспутываемый клубок! Ясно, что с такой матрицей просто так не поработаешь. Но! На протяжении нескольких лет ученые активно придумывают всё новые и новые модификации обычной ПЦР, существенно улучшающие результат.
Поскольку ГЦ-богатая матрица нуждается в более длительной и высокотемпературной денатурации, раньше пытались прогревать матрицу дополнительно, перед ПЦР. Однако, как можно догадаться, проблему это не решило. Еще в конце 90-х выяснили, что синтез ДНК прерывается на протяженных участках ЦГГ-повторов в присутствии K+, а чуть позже поняли, что виной тому те самые квадруплексы [12]. Поскольку в наиболее распространенный буфер для ПЦР как раз входит KCl, то самым очевидным решением было исключить его из состава буфера; это дало определенные результаты, но хотелось большего. Поэтому начали активно придумывать альтернативные буферы.
Сейчас часто проводят ПЦР с добавлением чистого Tris-HCl в качестве буфера. Tris –стандартный компонент для получения растворов нуклеиновых кислот: он дешев, и его буферные свойства высоки при рН 7–9 – значениях, физиологичных для живых организмов. В Tris обязательно добавляют хлорид магния в концентрациях, не ингибирующих ДНК-полимеразу и потому не уменьшающих выход специфического продукта. Очень часто смесь «улучшают» разными веществами, изменяющими свойства всей сложной системы ПЦР: ДМСО, бетаин, формамид, – они стабилизируют денатурированную ДНК, помогают снизить температуру плавления. Некоторые используют модифицированные дНТФ, в частности 7-деаза-дГТФ, и отмечают его эффективность (рис. 5); этот модифицированный нуклеотид препятствует формированию сложных дуплексов.

Рисунок 5. Усовершенствованная смесь для ПЦР – первое, что необходимо для амплификации (ЦГГ)n-области. Рисунок автора статьи.
Кроме компонентов смеси, существуют интересные варианты температурных циклов. Самый простой вариант, часто использующийся для амплификации не самых «труднопроходимых» последовательностей, – ПЦР с горячим стартом (hot-start PCR). Отличие этой модификации от стандартной ПЦР заключается в использовании специальных антител, предотвращающих активацию полимеразы до достижения нужной температуры, что позволяет избежать неспецифического синтеза. Для работы с экстремально ГЦ-богатыми матрицами (>83%) предложен вариант ПЦР под названием Slowdown (модификация Touchdown): медленные скорости нагрева и охлаждения, ступенчатое снижение температуры отжига через определенное количество циклов, добавление 7-деаза-дГТФ – всё это приводит к повышению выхода целевого продукта ПЦР.
Однако нельзя сказать, что проблема получения необходимых для дальнейшего анализа количеств ГЦ-богатых фрагментов (типа промоторной области FMR1) решена полностью: статьи на эту тему появляются часто, но опубликованные результаты противоречат друг другу; коммерческие компании соревнуются в разработке «волшебных» наборов, но стоить они могут настолько дорого, что их не в состоянии себе позволить даже успешные зарубежные лаборатории.
Определение размера (ЦГГ)n-области – это самый первый и очень важный этап в изучении синдрома ломкой X-хромосомы, который, однако, всё еще нуждается в оптимизации. Если мы научимся считать повторы быстро и качественно, то диагностика станет простой и относительно дешевой. Появится возможность проводить ее массово, а значит, отличать синдром ломкой X-хромосомы от множества других заболеваний, сопровождающихся умственной отсталостью, что крайне важно подбора терапевтических подходов.
Диагностика
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка – неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ – 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще – мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Генетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Патогенез
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской хромосоме. Этот ген кодирует производство протеина Е6АР, который представляет собой ферментный компонент сложной реакции деградации белков. Е6АР участвует в процессе образования убиквитина – белка системы протеасом, стимулирующего протеолиз дефектных белковых молекул в нейронах головного мозга. В норме убиквитин маркирует ненужные (неактивные, нефункциональные) белки с целью инициации их уничтожения. Е6АР обеспечивает закрепление убиквитина на поверхности молекулы белка-мишени. Потом протеасомы расщепляют его на пептидные остатки и на аминокислоты. При синдроме Ангельмана убиквитин не закрепляется на дефектных белках, они скапливаются в нервной ткани мозга, нарушается процесс синаптической передачи. Формируются отклонения, задержки в психическом и моторном развитии.
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Симптомы
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно нарастает задержка развития, ранее освоенные навыки сохраняются, но приобретение новых происходит медленно. Дети умеют сидеть, ползать, брать предметы и перекладывать их из руки в руку, поддерживать визуальный контакт, гулить и лепетать. Ходьба дается с трудом, нарушено чувство равновесия, наблюдаются частые падения, ушибы о мебель. Выявляется тремор и хаотичные движения конечностями, особенно руками. Речевые расстройства представлены как задержками, так и полным отсутствием экспрессивной речи. Дети либо совсем не говорят, либо используют лепет, простые слоги и слова общим объемом не более 10 единиц. Сохраняется понимание обращенной речи, стремление к общению, использование невербальных средств коммуникации: жестов, мимики, опосредованных знаков.
Основное поведенческое нарушение – гиперактивность. Дети часто веселятся и смеются без объективной причины, двигательно расторможены, неусидчивы, нецеленаправленны. У многих возникает патологическая привязанность к определенной игрушке или предмету быта, при появлении которого настроение сразу повышается, капризность и плач сменяются смехом. Концентрация внимания снижена, переключаемость быстрая и ненаправленная. Имеются трудности обучения, стойкое снижение интеллектуальных функций. Легко закрепляются стереотипии: раскачивание тела, размахивание руками. У 80% пациентов отмечается микроцефалия, недостаточный объем черепной коробки, эпилептическая активность мозга. Редко наблюдается снижение контроля движений языка, которое проявляется трудностями сосания груди или соски, последующим недостатком массы тела.
Характерные особенности внешности детей – косоглазие, сколиотическое искривление позвоночника, увеличение зубов и губ, разряжение зубного ряда, уплощение затылка, выступание вперед подбородка. Язык часто высунут, рот приоткрыт в улыбке. Развивается мышечная дистония, выраженность рефлексов сухожилий повышается, формируя специфичность моторики: пациенты ходят на прямых несгибающихся ногах, плечи приподнимают, руки сгибают в локтях. Своеобразный симптом – тяга к воде. Большинство детей чувствуют себя спокойнее в водной среде, им нравится плескаться в ванной, играть с корабликами в тазу.
Прогноз и профилактика
Выраженность симптомов синдрома Ангельмана может сильно различаться. Пациенты с легкими формами болезни имеют благоприятный прогноз: их речь становится более развернутой, улучшаются навыки самоконтроля при некоторых нарушениях двигательной сферы. При любой степени тяжести раннее начало и регулярное проведение медико-психологической помощи повышает качество жизни больных. Профилактика сводится к генетическому обследованию пар, в семьях которых есть ребенок с данным синдромом. Характер хромосомного дефекта (спорадический или наследственный) позволяет определить риск рождения второго больного ребенка.
Патогенез
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно – за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
Участок хромосом, подверженный структурным изменениям при наследственном синдроме Мартина-Белл, может находиться в четырех состояниях, характеризующихся различным удлинением повторяющихся последовательностей тринуклеотидов. При отсутствии болезни и носительства определяется нормальное количество повторов – от 6 до 39. В промежуточном состоянии диагностируется 40-60 повторов, в состоянии премутации – 55-200. В обоих случаях заболевание отсутствует. Поскольку экспансия тринуклеотидов возможна лишь в период гаметогенеза, премутация способна превратиться в полную мутацию. Это происходит при передаче измененного материнского гена, аллель «утяжеляется» во время овогенеза. При полной мутации выявляется больше 200 повторов ЦГГ, чаще всего – от 230 до 4 000.
Симптомы
Дети рождаются с увеличенной массой тела, в среднем – 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм – увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания – умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания – своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм – полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
Причины
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме материнского набора, но его характер и способ возникновения могут различаться. Иногда заболевание дебютирует в результате передачи измененной генетической информации от родителя, иногда является последствием спонтанных нарушений в геноме. Хромосомные аномалии удается определить примерно у 85-88% больных. Причиной синдрома может быть:
- Делеция. При данном дефекте часть генетического материала теряется или инактивируется. У 70% пациентов диагностируются обширные делеции области 15q12 хромосомы, в которой локализован активатор гена.
- Однородительская дисомия. ОРД определяется в 2-3% случаев болезни. В хромосомном наборе присутствуют две копии 15 хромосомы отца. Материнской хромосомы нет, ген также отсутствует.
- Дефект запечатления. Суть аномалии заключается в том, что центр запечатления, регулирующий активность локуса UBЕ3A, оказывается нефункциональным, «выключенным». Ген остается структурно целым, но не выполняет своих функций. Распространенность ДЗ – 3-5%.
- МутацияUBE3A. У 5-10% пациентов причиной болезни являются мутационные изменения гена. Они представлены инверсиями, микроделециями, транслокациями и дупликациями.
Синдром Мартина-Белл ( Синдром ломкой X-хромосомы )
Синдром Мартина-Белл – это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом – недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
Осложнения
Синдром Ангельмана – редкое малоизвестное заболевание. В связи с этим постановка диагноза и оказание медико-психолого-педагогической помощи зачастую проводятся несвоевременно, к 6-8 годам, что обуславливает низкую успешность коррекционных и лечебных мероприятий. Без физиотерапевтического лечения усугубляются нарушения опорно-двигательного аппарата – больные страдают от тяжелых форм сколиоза, самостоятельно передвигаются с трудом. Своеобразие внешности и поведения становится причиной ухудшения и без того затрудненной социальной адаптации. Все перечисленное влечет за собой утяжеление инвалидизации пациентов.
Общие сведения
Синдром назван по фамилии британского педиатра Г. Ангельмана. В 1965 году он первым описал симптомы заболевания и назвал его «синдромом счастливой марионетки», поскольку пациенты напоминали ему героя картины «Мальчик-марионетка». В те годы методы генетических исследований были еще не разработаны, установить причину патологии было невозможно. В 1987 году исследователи определили этиологию болезни и переименовали ее в синдром Ангельмана. Сейчас этот термин является официальным, но можно встретить синонимичные названия – «синдром марионетки», «синдром Петрушки», «синдром счастливой куклы». Распространенность составляет 1 случай на 10-20 тысяч новорожденных. Заболевание выявляется после первого года жизни (иногда – к 3-7 годам), чаще болеют мальчики.
Причины
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая – резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
Синдром Ангельмана ( Синдром марионетки , Синдром Петрушки , Синдром счастливой куклы )
Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития. Проявляется интеллектуальным отставанием, слабой сформированностью речи, навыков сидения и ходьбы, хаотичными движениями, гиперактивностью, симптоматической эпилепсией, беспричинным весельем и смехом, сколиозом, своеобразной походкой. Пациенты имеют особую внешность: рот крупный, зубы расположены редко, подбородок выдается вперед. Диагноз устанавливается на основании клинических данных, результатов генетического анализа. Специфическое лечение отсутствует, проводится симптоматическая терапия, оказывается психологическая и педагогическая помощь.
Общие сведения
Fragile x syndrome что это
Этиология и встречаемость синдрома ломкой Х-хромосомы. Синдром ломкой Х-хромосомы (MIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене FMR1 в Xq27.3. Синдром ломкой Х-хромосомы встречается с частотой 16-25 на 100 000 в общей популяции среди мужчин и в два раза реже среди женщин.
Синдром ломкой Х-хромосомы составляет 3-6% всех случаев умственной отсталости среди мальчиков с положительным семейным анамнезом по умственной отсталости при отсутствии врожденных пороков.
Патогенез синдрома ломкой Х-хромосомы
Продукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может сопровождать определенный подкласс мРНК от ядра к рибосомам.
Более 99% мутаций в гене FMR1 — экспансия нуклеотидного повтора (CGG)n в 5'-нетранслируемом участке гена. В нормальных аллелях FMR1 число повторов CGG составляет от 6 до приблизительно 50. В патогенных аллелях (или при полных мутациях) количество повторов более 200. Аллели с более чем 200 повторами CGG обычно имеют гиперметилированную последовательность повторов CGG и смежного промотора FMR1. Гиперметилирование инактивирует промотор FMR1, вызывая снижение экспрессии FMRP.
Полные мутации возникают из аллелей премутации (от 59 до 200 повторов CGG) с передачей мутантного аллеля FMR1 от матери (но не от отца); фактически при отцовской передаче премутации часто, наоборот, сокращаются. Полные мутации не могут возникать из нормальных аллелей. Поскольку длина неустойчивых повторов CGG увеличивается в каждом последующем поколении, если они передаются женщиной, обычно наблюдается увеличение числа пораженных потомков в последующих поколениях в семье; этот феномен называется генетической антиципацией.
Риск экспансии премутации в полную мутацию возрастает с увеличением числа повторов в премутации. Тем не менее не все премутации одинаково предрасположены к экспансии. Хотя премутации встречаются сравнительно часто, переход в полную мутацию наблюдают только в ограниченном количестве гаплотипов, т.е. когда есть склонность гаплотипа к экспансии.
Эта склонность гаплотипа частично может быть связана с присутствием нескольких триплетов AGG, вставленных в последовательность повторов CGG; оказывается, такие триплеты AGG тормозят экспансию повторов CGG, следовательно, их отсутствие в некоторых гаплотипах может предрасполагать к экспансии.
Фенотип и развитие синдрома ломкой Х-хромосомы
Синдром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее пораженные индивидуумы также имеют поведенческие аномалии, включая гиперактивность, размахивание руками, истерики, плохой зрительный контакт и признаки аутизма. Физические характеристики мужчин изменяются с пубертатом.
До полового созревания пораженные мальчики имеют несколько увеличенный размер головы и некоторые другие неотчетливые симптомы; после наступления половой зрелости у них частые более отчетливые признаки (длинное лицо с выдающейся челюстью и лбом, крупные ушные раковины, макроорхидизм).
Поскольку эти клинические признаки не уникальны для синдрома ломкой Х-хромосомы, диагноз зависит от молекулярного обнаружения мутаций. Пациенты с синдромом ломкой Х-хромосомы имеют нормальную продолжительность жизни.
Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа зависит от мозаицизма метилирования повторов и их числа. Поскольку полные мутации неустойчивы, некоторые пациенты имеют смесь клеток с числом повторов, колеблющимся от премутации до полной мутации (мозаицизм числа повторов).
Все мужчины с мозаицизмом числа повторов больны, но часто имеют более высокие показатели умственного развития, чем пациенты с полной мутацией в каждой клетке; у женщин с мозаицизмом числа повторов клинические проявления варьируют от нормы до полного проявления. Аналогично некоторые пациенты имеют смесь клеток с метилированием повторов CGG и без него (мозаицизм метилирования повторов). Все мужчины с мозаицизмом метилирования больны, но часто имеют более высокие показатели умственного развития, чем с гиперметилированием в каждой клетке; женщины с мозаицизмом метилирования также могут быть здоровыми или больными.

Очень редко пациенты имеют полную мутацию, неметилированную во всех клетках; независимо от пола, степень тяжести у них варьирует от нормы до полной клиники. Кроме того, у женщин фенотип зависит от степени смещения инактивации Х-хромосомы.
Носительницы премутации (но не полных мутаций) имеют 20% риск ранней дисфункции яичников. Мужчины-носители премутации имеют риск развития синдрома FXTAS. FXTAS проявляет себя как поздняя прогрессирующая мозжечковая атаксия с интенционным тремором. У больных могут также присутствовать снижение краткосрочной памяти и двигательных функций, когнитивные нарушения, а также паркинсонизм, периферическая нейропатия, проксимальная мышечная слабость нижних конечности и дизавтономия.
Пенетрантность FXTAS зависит от возраста, обнаруживается в 17% в течение шестого десятилетия жизни, в 38% в течение седьмого десятилетия, в 47% в течение восьмого десятилетия и в трех четвертях старше 80 лет. FXTAS может встречаться и у некоторых женщин — носительниц премутации.
Особенности фенотипических проявлений синдрома ломкой Х-хромосомы:
• Возраст начала: детство
• Умственная недостаточность
• Дисморфическое лицо
• Постпубертатное увеличение яичек у мужчин (макроорхидизм)
Лечение синдрома ломкой Х-хромосомы
К настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и фармакологическое лечение поведенческих проблем.
Риски наследования синдрома ломкой Х-хромосомы
Риск того, что женщина с премутацией будет иметь больного ребенка, определяется размером премутации, полом плода и семейным анамнезом. Эмпирически риск для носителя перестройки иметь больного ребенка может достигать 50% для каждого мальчика и 25% для каждой девочки, но зависит от размера премутации. На основе анализа сравнительно небольшого количества матерей-носительниц известно, что риск повторения может снижаться, если премутация уменьшается со 100 до 59 повторов. Пренатальная диагностика доступна за счет использования ДНК плода из ворсин хориона или амниоцитов.
Пример синдрома ломкой Х-хромосомы. Р.Л., 7-летний мальчик, направлен в клинику педиатрии в связи с умственной задержкой и гиперактивностью. Он не смог посещать детский сад, поскольку был агрессивным, не в состоянии выполнять задания, имел бедные речевые и двигательные навыки. Несмотря на задержанное развитие, он не потерял основных этапов: сидел к 10-11 мес, ходить начал в 20 мес, говорил два или три ясных слова в 24 мес.
В остальном ребенок здоров. Его мать и тетя по матери имели небольшие проблемы обучения в детстве, дядя по матери умственно задержан. Данные медицинского осмотра в норме, за исключением гиперактивности.
Врач рекомендовал несколько тестов, включая кариотипирование, функциональные исследования щитовидной железы и ДНК-анализ на синдром ломкой Х-хромосомы. Анализ гена FMR1 методом блот-гибридизации по Саузерну соответствовал синдрому ломкой Х-хромосомы.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
МКБ-10
МКБ-10
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Диагностика
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и генетики. Родители предъявляют жалобы на отсутствие речи, двигательные стереотипии, трудности формирования ходьбы и других двигательных навыков, гиперактивность. Проводится дифференциальная диагностика, в ходе которой должны быть исключены более распространенные заболевания, такие как умственная отсталость, расстройства аутистического спектра, деменции, мутизм, несимптоматические формы эпилепсии. Комплексное исследование включает следующие процедуры:
- Общий осмотр. На наличие синдрома часто указывают специфические черты внешности пациентов: высунутый язык, слюнотечение, крупный рот, широкие редкие зубы, выступающая вперед нижняя челюсть, плоская форма затылка. Характерен светлый оттенок кожи, глаз и волос. Походка детей напоминает движения куклы-марионетки из-за усиления сухожильных рефлексов и снижения мышечного тонуса.
- Осмотр психиатром. В 100% случаев синдрома Ангельмана диагностируется выраженная задержка в развитии психики, отсутствие самостоятельной речи или очень скудный словарный запас. Коммуникация осуществляется с помощью мимики, жестов, рисунков. В поведении отмечается гиперактивность, стереотипные движения руками, беспричинный смех.
- Неврологическое обследование. У всех пациентов определяется атаксия и тремор конечностей. 80% больных имеют постнатальную микроцефалию – окружность головы новорожденного меньше 32 см, к 12 месяцам – около 42 см. По данным ЭЭГ выявляется симптоматическая эпилепсия (высокоамплитудные разряды медленных комплексных волн), клинически возможны судорожные припадки. У некоторых детей имеется косоглазие, диффузное снижение мышечного тонуса, усиление сухожильных рефлексов, гиперкинезы.
- Генетическое исследование. Лабораторная диагностика нацелена на выявление мутаций и делеций в гене UBE3A. Последовательно проводится комплекс процедур, включающий флуоресцентную гибридизацию in situ, анализ мутации центра запечатления, метилирование ДНК СА/ПВС региона, диагностику делеции методом микроматричного анализа, поиск мутационных изменений в локусе UBE3A.
Читайте также: